

Chronic liver disease and/or portal hypertension may be associated with one of the two pulmonary vascular complications: portopulmonary hypertension and hepatopulmonary syndrome. These pulmonary vascular disorders are notoriously underdiagnosed; however, they have a substantial negative impact on survival and require special attention in order to understand their diagnostic approach and to select the best therapeutic options. Portopulmonary hypertension results from excessive vasoconstriction, vascular remodeling, and proliferative and thrombotic events within the pulmonary circulation that lead to progressive right ventricular failure and ultimately to death. On the other hand, abnormal intrapulmonary vascular dilations, profound hypoxemia, and a wide alveolar-arterial gradient are the hallmarks of the hepatopulmonary syndrome, resulting in difficult-to-treat hypoxemia. The aim of this review is to summarize the latest pathophysiologic concepts, diagnostic approach, therapy, and prognosis of portopulmonary hypertension and hepatopulmonary syndrome, as well as to discuss the role of liver transplantation as a definitive therapy in selected patients with these conditions.
La hipertensión portal con o sin hepatopatía crónica puede estar asociada a complicaciones en la vasculatura pulmonar, en concreto al desarrollo de hipertensión portopulmonar y al síndrome hepatopulmonar. Dichas entidades nosológicas son claramente subdiagnosticadas; sin embargo, confieren un impacto negativo en la sobrevida y el pronóstico de estos enfermos, requiriendo especial atención, con el fin de establecer una estrategia adecuada en cuanto al abordaje diagnóstico y las implicaciones y conductas terapéuticas a seguir ante su detección. La hipertensión portopulmonar resulta de una remodelación vascular pulmonar que incluye fenómenos vasoconstrictivos, proliferativos y protrombóticos en la circulación pulmonar, llevando a la insuficiencia ventricular derecha progresiva y a la muerte. Por otro lado, las dilataciones vasculares intrapulmonares anormales, la hipoxemia significativa y el incremento en el gradiente alvéolo-arterial caracterizan al síndrome hepatopulmonar, siendo un reto terapéutico el manejo de la hipoxemia refractaria. Esta revisión se enfocará a sintetizar los aspectos fisiopatológicos, el abordaje diagnóstico y terapéutico, así como las consideraciones pronósticas de la hipertensión portopulmonar y del síndrome hepatopulmonar, además de describir el papel del trasplante hepático como terapia definitiva en pacientes seleccionados.
Chronic liver disease and/or portal hypertension are leading causes of morbidity and mortality among the Western adult population and have been associated with two particular pulmonary vascular complications, portopulmonary hypertension (POPH) and hepatopulmonary syndrome (HPS), resulting in significant morbidity and mortality, particularly in the setting of end-stage liver disease requiring liver transplantation (LT). In the last decade there has been increasing awareness and recognition of these entities, especially when LT is being considered, influencing the pre-, trans- and post-transplant outcomes. HPS is characterized by intrapulmonary vascular dilatation and hypoxemia, while POPH is characterized by increased pulmonary vascular resistance (PVR) and mean pulmonary arterial pressures (mPAP).1 This review will focus on the relevant clinical aspects of both POPH and HPS, providing the clinician with the tools to guide him in the care of this complex subset of patients.
Portopulmonary hypertension (POPH)DefinitionPOPH is best defined as a condition characterized by increased mean pulmonary arterial pressure (mPAP) and pulmonary vascular resistance (PVR), resulting in pulmonary arterial hypertension (PAH) in association with portal hypertension, whether or not portal hypertension is related to underlying chronic liver disease or not.1,2 At the 2008 Dana Point World symposium on pulmonary hypertension (PH), POPH was classified within the World Health Organization (WHO) PH group 1, becoming part of one of the associated forms of PAH.3,4 Currently, specific hemodynamic criteria exist for the accurate definition and diagnosis of POPH. These hemodynamic parameters require performing a right heart catheterization in which the following measurements are obtained: mPAP>25mmHg at rest, PVR>240dynes/s/cm−5 (or >3 Woods units; 1 Wood unit=80dynes/s/cm−5), and a mean pulmonary arterial occlusion pressure (mPAOP)<15mmHg or a transpulmonary gradient (TPG, the difference between mPAP and mPAOP)>12mmHg.5 TPG sometimes can be helpful for clinicians when the assessment of PVR and/or the mPAOP do not meet the current criteria. We believe that understanding the cardiopulmonary hemodynamics and physiology in patients with portal hypertension and liver disease is of paramount importance, since, in general, patients with advanced liver disease suffer from hyperdynamic cardiovascular system, characterized by high cardiac output (CO) and low peripheral vascular resistance and arterial hypotension, as well as increased blood volume due to fluid and electrolyte imbalances typically seen in advanced cirrhosis. Hemodynamically this results in elevated mPAP, CO, and mPAOP with a normal or low PVR. Interestingly, approximately 30–50% of patients with chronic liver disease have decreased systemic vascular resistance, low PVR and elevated CO, making the elevations in mPAP due to the increased CO, leading some investigators and researchers to propose a new cutoff for PVR>120dynes/s/cm−5 to be used as a novel definition of POPH.6,7
EpidemiologyThe incidence and prevalence of POPH is difficult to assess and is not well defined. POPH is a relatively rare condition; however, most commonly observed among patients being evaluated for LT, where it has been reported in 6–8% of the patients with end-stage liver disease.8,9 In the French national registry for pulmonary hypertension (PH), patients with POPH accounted for 10% of the PAH population, just behind idiopathic PAH (iPAH), PAH associated with congenital heart disease and PAH associated with connective tissue disorders.10 Reports on the prevalence of POPH have varied greatly over the last ten years, which may be the reflection of changing clinical practices by adopting a better screening and diagnostic work-up, particularly in patients undergoing LT evaluation.11
POPH is commonly diagnosed 4–7 years after the presence of portal hypertension has been established, and is most commonly described during the fifth decade of life.12 Kawut et al.13 performed an observational prospective cohort study to evaluate the risk factors for the development of POPH, and found that female sex was associated with a higher risk for POPH than male sex, and also autoimmune hepatitis as etiology of liver disease was more commonly associated to POPH than other etiologies, whereas chronic hepatitis C infection was less likely to be associated with POPH, concluding that hormonal and immunologic factors could play an important role for the development of POPH. Krowka et al.14 evaluated the survival and hospitalization rates in the Registry to Evaluate early and long-term PAH disease management (REVEAL registry) of patients with POPH when compared with iPAH; finding that POPH patients had significantly poorer survival rates compared with iPAH, of note, POPH patients were less likely to be in PAH-specific vasodilator therapy. Interestingly, the severity of chronic liver disease as estimated by the Child-Pugh score or the Model for End-stage Liver Disease (MELD) score were not associated with the presence or severity of POPH.9,13 Le Pavec et al.15 retrospectively analyzed the charts of patients referred to the French referral center for PAH, evaluating 154 patients, finding that the presence and severity of cirrhotic liver disease and cardiac index were the major independent prognostic factors for survival.
PathophysiologyPOPH is pathobiologically indistinct when compared with other forms of PAH. The pulmonary endothelium is a complex system and the pathogenesis of POPH is not completely understood; however, the vascular remodeling that takes place in POPH is likely an end-product of the complex interaction and dysregulation of numerous vasoactive, proliferative, angiogenic and prothrombotic signals and modulators.1,2 Most of the theories to try to explain the pathophysiology of POPH have been derived from recent advances in the cellular and molecular studies on iPAH. A significant up-regulation of the potent pulmonary vasoconstrictor and proliferator endothelin-1 (ET-1) has been found in patients with POPH, and is thought likely to result from the impaired clearance or increased production of ET-1 by the deranged liver.16 Down-regulation or decreased levels of prostaglandin I-2 (prostacyclin) synthase have also been found in patients with PAH, including those with POPH.17
A proposed mechanism for the development of POPH includes the initial and constant shear stress and increased blood flow (high cardiac output status) in liver disease, activating a cascade of events that may eventually lead to pulmonary endothelial dysfunction and pulmonary vascular remodeling.18 Another hypothesis that has been proposed is that toxic products from the digestive tract that are normally metabolized and cleared in the liver bypass the liver via portosystemic shunts, directly damaging the pulmonary vascular endothelium.19 The development of portosystemic shunts, a decrease in liver phagocytic capacity, and increased frequency of bacterial translocation facilitate the expression and activation of pro-inflammatory cytokines, pro-angiogenic factors and bacterial endotoxins which probably induce pulmonary endothelium damage and dysfunction.20 These changes ultimately lead to increased PVR and increased right heart workload, which then result in the changes seen at the time of right heart catheterization as described earlier.
DiagnosisMost patients with POPH are asymptomatic at the time of diagnosis, and hence a high index of suspicion is needed to identify this condition in patients with cirrhotic or non-cirrhotic portal hypertension. When symptoms are present, they generally include dyspnea on exertion, fatigue and in more severe cases, chest pain, orthopnea, hemoptysis and syncope.21,22 On physical examination patients can present with a loud pulmonic valve closure (P2), split second heart sound, and tricuspid insufficiency murmur and right ventricular heave. Also common are signs of right heart failure such as increased jugular venous pressure, right-sided S3 or S4 gallop, lower extremity edema and pulsatile liver.22 The presence of symptoms and/or signs of pulmonary hypertension in a patient with portal hypertension should lead the clinician to consider POPH in the differential diagnosis and to embark in further diagnostic evaluation. The main differential diagnoses include idiopathic PAH, PAH associated with collagen vascular disease, thromboembolic disease, infection with human immunodeficiency virus (especially in patients with chronic viral hepatitis B or C), or PAH associated with primary cardiac or lung diseases.23 The clinician should also consider other causes of cardiopulmonary symptoms in the cirrhotic patient, including ascites, hepatic hydrothorax, sarcopenia and muscle waisting, deconditioning, anemia and hepatopulmonary syndrome (HPS).24
After a thorough history and physical exam, the most useful tool to aid in the diagnosis of POPH is transthoracic echocardiography (TTE), with a sensitivity >95% and specificity close to 80–90%.25,26 The parameter used to best assess pulmonary pressure is the right ventricular systolic pressure (RVSP) or pulmonary artery systolic pressure (PASP).26 TTE can also provide detailed information on the function of the right ventricle that is complementary to the RVSP measurement and improves our ability to determine the clinical consequences of POPH in a specific patient.27 Of note, up to 20–30% of patients undergoing TTE do not have tricuspid regurgitation and hence RVSP cannot be estimated. Fortunately, most of these patients will not have pulmonary hypertension, and hence no further work-up is necessary.9
A RVSP cutoff of 30–50mmHg has been used by different institutions to select patients who require further diagnostic evaluation.9,28 Although several non-invasive modalities have been evaluated, the current diagnostic “gold-standard” remains right heart catheterization. The hemodynamic measurements at the time of right heart catheterization are indispensable to differentiate increases in mPAP from a hyperdynamic state or from volume overload, both common in the cirrhotic patient, from true POPH, which will by definition have not only a mPAP>25mmHg but also a PVR>240dynes/s/cm−5 and a mPAOP<15mmHg or a TPG>12mmHg, as described earlier.2
In the recent past, several research groups have attempted to improve our ability to diagnose POPH using non-invasive methods. Tsiakalos et al. evaluated the serum levels of endothelin (ET) 1, 2 and 3 and described a significant increase in ET-1 and ET-1 to ET-3 ratio in patients with POPH as defined by an RVSP>40mmHg compared to patients with normal RVSP.29 Pellicelli et al. showed significantly increased levels of serum ET-1 and interleukin 6 among patients with POPH in comparison to cirrhotic patients with or without hyperdynamic circulation diagnosed by right heart catheterization.30 Potentially more useful in clinical practice are the findings of Bernal et al. on the use of N-terminal natriuretic peptide type B (NT-proBNP) in patients with cirrhosis and POPH. They were able to demonstrate increased serum levels of NT-proBNP in patients with mPAP>25mmHg on right heart catheterization compared to patients with lower mPAP (1950±1245 vs. 1407±945, P=0.04) and demonstrated a weak correlation between NT-proBNP levels and severity of pulmonary hypertension (mPAP) on right heart catheterization.31,32 In the future it might be possible to accurately distinguish cirrhotic patients with POPH vs. those with other causes of elevated RVSP on TTE without the need for right heart catheterization, but for now this invasive and costly diagnostic test continues to be the gold-standard of diagnosis and monitoring of therapy for PAH.
TherapyPOPH is a progressive and lethal disease. If left untreated, moderate to severe POPH has a mortality rate of 54% within 1 year and a dismal 14% survival rate at five years.33 Conversely, if treated successfully, a 45% survival rate at 5 years can be expected.33 Despite having better hemodynamics measurements at baseline and better hemodynamic response to therapy, POPH fairs worse than iPAH. This suggests that factors other than pulmonary hemodynamics participate in the poor long-term survival of these patients.33,34 Recently, Talwalkar et al. have demonstrated that large portosystemic shunts defined as a greater than 10mm in diameter were associated with poor response to treatment in 68% of study patients with POPH.35
The role of specific therapies for POPH remains unclear since a therapeutic approach has never been scientifically evaluated. Nonetheless, as POPH is classified within the WHO group 1 PAH,36 therapies that have been FDA approved to treat PAH have been considered adequate to treat POPH.
The main goal of therapy in POPH is to improve exercise tolerance, quality of life, reduce symptoms and reach a hemodynamic response that will facilitate LT when indicated.37 The usual approach to treating POPH is to initiate therapy when mPAP is >35mmHg on right heart catheterization, with a final goal to reduce mPAP to ≤ 35mmHg and PVR to <400dynes/s/cm−5, so patients can be considered for liver transplantation, as further discussed in the corresponding section.
POPH is treated with the same general measures that are used to treat iPAH.38 The aim of these therapies is to unload the right ventricle and prevent right ventricular failure. Diuretics, supplemental oxygen, low salt diet, weight control, routine influenza and pneumonia immunizations, mild to moderate exercise and participation in local support groups for PAH are useful to improve quality of life and improve symptoms. The use of calcium channel blockers, beta blockers and anticoagulation are not recommended in POPH.1 The calcium channel blockers have the potential of producing mesenteric vasodilatation that may worsen portal hypertension.39 Beta blockers with their negative inotropic and chronotropic effects worsen exercise capacity and pulmonary hemodynamics.40 Although chronic oral anticoagulation is indicated in iPAH, it is not generally indicated in POPH due to the high risk of bleeding associated with the presence of esophageal varices.
In the past 15 years, new targeted therapies have been approved by the FDA to treat PAH. Three different groups of medications are currently available: prostacyclin analogs, endothelin receptor antagonist (ERAs) and phosphodiesterase-5 inhibitors (PD-5).
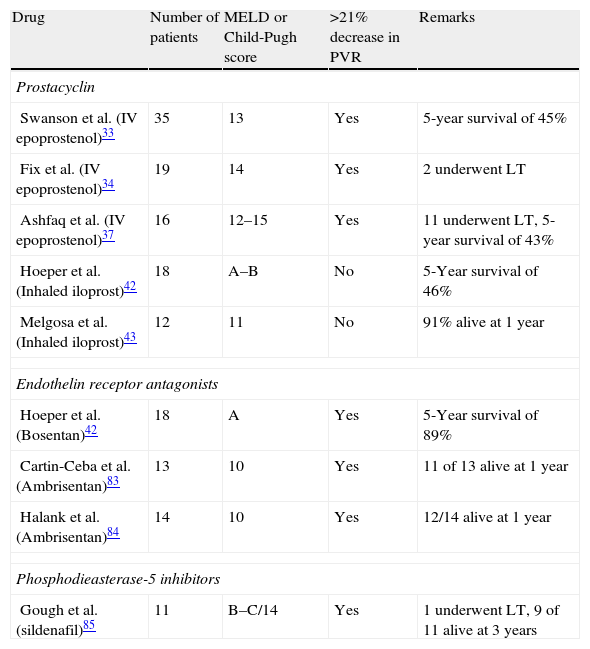
Although randomized trials have never been performed in POPH, several case series and retrospective studies have shown substantial response to therapy; as defined by a >20% decline in PVR after 12 weeks of treatment.41 Furthermore, these studies revealed improvements in exercise tolerance and overall survival, as well as a decrease in mPAP to values where LT is possible (Table 1). Of note, inhaled prostacyclin does not improve long-term hemodynamics when used as monotherapy.42,43 Up to now, IV or SQ prostacyclin has been considered the drug of choice to treat POPH with the intention of future liver transplantation. Unfortunately, only few patients may be able to reach an mPAP that will allow liver transplantation. In addition, IV/SQ prostacyclin is delivered at a high financial cost and at high risk of complications and side effects. Since therapies other than prostacyclin offer similar benefits, a plausible therapeutic intervention calls for initiation of a goal oriented therapy as described by Hoeper et al. with a minor protocol modification.44 Patients will need a follow-up right heart catheterization to re-evaluate mPAP and PVR. The protocol calls for regular clinical follow-up at 1–6 months with interval measurement of the 6-min walk test, functional assessment of dyspnea and hemodynamic data at 3–6 months of therapy. Treatment can ideally start with an oral agent, preferably ERAs, since the hemodynamic improvement of PD-5 seems to plateau after 3 months. After the addition of a second oral agent, the addition of a third medication will depend on the clinical and/or hemodynamic response and the patient's candidacy for LT. This protocol may reduce the need for prostacyclin treatment and better select those patients who will benefit from liver transplantation.
Current specific medical therapies for portopulmonary hypertension.
| Drug | Number of patients | MELD or Child-Pugh score | >21% decrease in PVR | Remarks |
| Prostacyclin | ||||
| Swanson et al. (IV epoprostenol)33 | 35 | 13 | Yes | 5-year survival of 45% |
| Fix et al. (IV epoprostenol)34 | 19 | 14 | Yes | 2 underwent LT |
| Ashfaq et al. (IV epoprostenol)37 | 16 | 12–15 | Yes | 11 underwent LT, 5-year survival of 43% |
| Hoeper et al. (Inhaled iloprost)42 | 18 | A–B | No | 5-Year survival of 46% |
| Melgosa et al. (Inhaled iloprost)43 | 12 | 11 | No | 91% alive at 1 year |
| Endothelin receptor antagonists | ||||
| Hoeper et al. (Bosentan)42 | 18 | A | Yes | 5-Year survival of 89% |
| Cartin-Ceba et al. (Ambrisentan)83 | 13 | 10 | Yes | 11 of 13 alive at 1 year |
| Halank et al. (Ambrisentan)84 | 14 | 10 | Yes | 12/14 alive at 1 year |
| Phosphodieasterase-5 inhibitors | ||||
| Gough et al. (sildenafil)85 | 11 | B–C/14 | Yes | 1 underwent LT, 9 of 11 alive at 3 years |
All FDA approved therapies for PAH increase cardiac output and have the potential of worsening portal hypertension and inducing hypersplenism by increasing the hepatic venous pressure gradient. Although described with prostacyclin analogs,45 this particular situation is rare but more frequent than in iPAH. In regards to the increase in hepatic venous pressure, only Melgosa et al. have studied the acute and chronic hemodynamic responses to inhaled prostacyclin. They demonstrated that the acute and long-term effect of inhaled prostacyclin produces rapid and selective pulmonary vasodilatation without altering the hepatic hemodynamics.43 Patients with POPH generally die from PH related problems with right-heart failure, progression of their liver disease, infectious complications and sepsis, and from hepatocellular cell carcinoma.
POPH and liver transplantationBecause of its mostly asymptomatic presentation and its significant consequence to clinical outcomes after LT, all patients with cirrhosis being considered for transplantation should be screened for POPH. Due to its usefulness as a screening tool the American Association for the Study of Liver Diseases (AASLD) recommends that all patients evaluated for liver transplantation undergo TTE and that if RVSP is elevated, further evaluation with right heart catheterization is warranted.46 Some authors recommend periodically screening patients on the transplant wait list with TTE to look for new cases of POPH, as it can at times develop quite rapidly and may not be present on initial transplant evaluation.11
A right heart catheterization provides not only an accurate diagnosis of POPH, it also allows the assessment of the severity of POPH, which has prognostic implications in LT. Patients with mild POPH (mPAP of 25–34mmHg) have similar perioperative and postoperative morbidity and mortality after LT than patients without POPH and can proceed to LT without further intervention, while patients with moderate POPH (mPAP of 35–44mmHg) have a high risk of perioperative and post-transplant mortality and should be targeted for therapy to reduce mPAP to <35mmHg prior to LT (see section on therapy). Patients with severe POPH (mPAP≥45mmHg) should be excluded from LT as it results in an exceedingly high mortality rate.47
As POPH does not always resolve after transplantation, it is not considered as an indication for LT. This is different from HPS, which clearly improves after LT and is one of the indications for transplant. Although POPH has been shown to resolve after LT in some patients, it has also been described that others require long-term pulmonary vasodilator therapy after LT and that yet others suffer from worsening PAH after LT and there are even case reports of PAH developing “de-novo” after transplant. Nonetheless, in the United States the United Network for Organ Sharing (UNOS) allows patients with mild to moderate POPH be listed for LT and provides MELD exception points (MELD score=26) if the LT candidate satisfies the following criteria: (a) confirmed POPH by right heart catheterization with mPAP>35mmHg; (b) at least 12 weeks of approved PAH therapy that results in mPAP<35mmHg and PVR<400dynes/s/cm−5 with a satisfactory right ventricular function as assessed by the individual transplant center. If the patient is not transplanted within a 6-month period, additional MELD exception points may be granted after approval by the regional review board.48 Patients with moderate POPH refractory to medical therapy or with severe POPH are routinely excluded from LT due to the very high mortality after LT. Recently though, cases of combined heart, lung and liver or combined lung and liver transplant have been described to address this specific issue. Clearly this should only be undertaken by very experienced centers in highly selected patients.49,50
Hepatopulmonary syndromeDefinitionCardiovascular abnormalities are present in most patients with advanced liver disease. The term “hepatopulmonary syndrome” (HPS) was coined by Kennedy and Knudson in 1977.51 This syndrome is characterized by defects in arterial oxygenation as a consequence of shunting through pulmonary vascular dilatations in the setting of liver disease,5,52 and has three classical components: liver disease, intrapulmonary vascular dilatations (IPVD), and poor oxygenation. The oxygenation defect involves a widened aged-corrected alveolar-arterial oxygen gradient (P[A-a]O2) at room air, with or without concomitant hypoxemia.5
HPS may present in patients with or without portal hypertension in the setting of acute or chronic liver disease. Indeed it may be present in patients with acute liver failure. There is only a weak association between the severity of the liver disease and the degree of hypoxemia and the cause of liver disease does not seem to be related with the development of HPS.53 HPS may occur in patients with other comorbidities, including cardiopulmonary conditions.54 Of relevance is the fact that HPS increases morbidity and mortality among patients with end-stage liver disease awaiting LT.55
In HPS hypoxemia is defined on the basis of an increased alveolar-arterial oxygen gradient, P[A-a]O2≥15mmHg in adults or P[A-a]O2≥20mmHg in patients older than 64 years of age, at sea level, and at room air (FiO2 21%). This method is the most sensitive to detect early deoxygenation, even before arterial oxygen tension (PaO2) becomes abnormally low.5 However, the presence of hypoxemia is not sufficient to define HPS, it must be accompanied by IPVD (defined as increased pulmonary capillary diameter ranging from 15 to 60μm) as assessed by the presence of an abnormal contrast-enhanced echocardiography (CEE) and/or perfusion lung scanning with technetium-99m-labeled macroaggregated albumin (99 TcMAA).
Pathogenesis and pathophysiologyThe main pathologic features of HPS include gross dilatation of the pre-capillary and capillary pulmonary vessels (to 15–100μm in diameter), pleural and pulmonary arterio-venous shunts and portopulmonary venous anastomoses. The above vascular abnormalities result in impaired oxygenation of venous blood as it passes through the pulmonary circulation. The pulmonary vascular dilatation results in the passage of mixed venous blood either rapidly or even directly, through intrapulmonary shunts, into the pulmonary veins. The defect in oxygenation is due to a ventilation-perfusion mismatch characterized by increased blood flow while alveolar ventilation is preserved; this phenomenon is enhanced by the absence or impairment of hypoxic pulmonary vasoconstriction.18
Experimental and clinical data suggest that increased production of nitric oxide (NO) in the lungs plays a central role in the pathogenesis of HPS. This potent vasodilator has been linked to vascular abnormalities. Increased intrapulmonary levels of NO have been described in cirrhotic patients with HPS,56 and their subsequent normalization after transplantation were associated to resolution of HPS.57,58 Increased expression of pulmonary endothelial-cell endothelin B receptors (ETB) and increased hepatic production and release of ET-1 contribute to an increase in eNOS (endothelial Nitric Oxide Synthase) expression and enhanced NO production in an animal model of HPS.5,18
Recently, angiogenesis has been recognized as a relevant contributor to the pathophysiology of HPS in animal models, and is mediated by the activation of vascular endothelial grow factor A produced in part by activated intravascular monocytes.59 In humans, several polymorphisms in the genes involved in the regulation of angiogenesis have been associated with the risk of HPS (CAV 3, ENG, NOX4, ESR2, VWF, RUNX1, COL18A1, COL18A1 and TI31).60,61
Clinical manifestationsHPS is typically diagnosed in patients with cirrhosis who present with shortness of breath, cyanosis, finger clubbing and clinically significant hypoxemia (PaO2<60mmHg), but the majority of patients with HPS are either asymptomatic or develop insidious onset of dyspnea. Dyspnea on exertion, at rest, or both is the predominant presenting symptom; however, this symptom is a nonspecific finding, and there are no hallmark signs on physical examination.
In addition, hypoxemia may worsen (PaO2 arterial blood decreased ≤5% or 4mmHg) when the patient moves from supine to an upright position (orthodeoxia), which produces the clinical symptom of platypnea, or worsening shortness of breath on standing. This clinical feature is present in up to 25% of patients with HPS. This clinical phenomenon is a reflection of increased perfusion of the lung bases, where vascular dilatation predominates in HPS, leading to worsening oxygen exchange, reduced PaO2 and the increased feeling of shortness of breath.5,18,62
Pulmonary function test typically demonstrate a reduced carbon monoxide diffusion capacity in the presence of normal spirometric volumes, but this finding is not specific for HPS.62
Noticeably, there is no correlation between the severity of HPS and the severity of the liver disease, on basis of different models of liver disease severity (i.e. Child-Pugh class or MELD score).62
Clinicians should also be aware that common chronic pulmonary diseases (i.e. COPD, asthma, etc.) can coexist in a third of patients with HPS.5,55 Extrapulmonary complications associated with right to left pulmonary shunts are rare and include brain abscess and intracranial hemorrhage.63,64 The physician must also consider the syndrome of hereditary hemorrhagic telangiectasia (HHT or Rendu-Osler-Weber syndrome) in the differential diagnoses of HPS, as it shares some similarities, and both can be associated with severe hypoxemia caused by pulmonary vascular dilatation, which may be diffuse or discrete in nature.
Staging of severityThe severity of HPS not only directly influences patient survival, it also helps the clinician make decisions regarding timing of LT and the risk of mortality associated with this procedure. This grading system is based on oxygenation abnormalities in four stages, as depicted in Table 2. As these oxygenation abnormalities become more severe, much more so is their impact on the patients’ quality of life and the more clinical symptoms are apparent. The severity of HPS should also prompt the physician to pursue specific therapeutic interventions (e.g. long term oxygen therapy and embolotherapy) to improve or reverse some of the symptoms and limitations associated with HPS.65,66
Grading of severity of hepatopulmonary syndrome.
| Stagea | P[A-a]O2 (mmHg)b,d | PaO2 (mmHg)c |
| Mild | ≥15 | ≥80 |
| Moderate | ≥15 | <80 to ≥60 |
| Severe | ≥15 | <60 to ≥50 |
| Very Severe | ≥15 | <50 (<300 on 100% O2) |
P[A-a]O2: alveolar-arterial oxygen tension difference, the abbreviated formula for alveolar-arterial gradient is as follows: PAO2–PaO2=(FiO2[Patm–PH2O]−[PaCO2/0.8])−PaO2, where PAO2 denotes partial pressure of alveolar oxygen, PaO2 partial pressure of arterial oxygen, FiO2 fraction inspired oxygen, Patm atmospheric pressure, PH2O partial pressure of water vapor at body temperature, and PaCO2 partial pressure of arterial carbon dioxide. PaO2: arterial oxygen tension.
aAll with positive contrast-enhanced echocardiography.
The prevalence of HPS in patients with cirrhosis and end-stage liver disease ranges from 4% to 30%,67 and the differing prevalence is primarily due to heterogeneity of the applied diagnostic criteria.
Poor quality of life and increased mortality have been described in cirrhotic patients with HPS, and survival is significantly decreased compared with cirrhotic patients without HPS.68 The presence of HPS worsens the prognosis of patients with liver disease, regardless of liver transplant status. Studies in patients who were not candidates for liver transplant have shown a median survival of 87 months and a 5-year survival of 63%, while patients with HPS had a median survival of 24 months and a 5-year survival of 23%.69 Even after LT, patients with HPS tend to do worse than patients without HPS, even in those patients with improved post-transplant arterial oxygenation (PaO2>50),70 with high postoperative mortality, mostly within the first 3 months after surgery. In a large retrospective study, the 1- and 4-year morality was 38.5% and 42.3%, compared with patients without HPS, who had 1- and 4-year mortality rates of 0% and 20%, respectively.71 In spite of these findings, LT does alter the natural course of HPS, and thus has become the treatment of choice for HPS in many centers. Certainly, in selected cases, successful LT has resulted in complete resolution of HPS in the majority of survivors of the early post-surgical period.18
The causes of death associated with HPS are usually multifactorial and related to the complications of liver disease (e.g. hepatic failure, multisystem organ failure due to sepsis, hepatocellular carcinoma, gastrointestinal bleeding), much more so than pulmonary complications. Although oxygenation usually worsens over time (mean decline of 5.2mmHg per year), it is not common for severe hypoxemic respiratory failure to be the primary cause of death.18
DiagnosisThe diagnosis of HPS requires the presence of abnormal arterial gas exchange associated with evidence of IPVD, as described in the section on “definition of HPS”.
A recent prospective study, using an oxygen saturation (SpO2) threshold value of <95% was highly sensitive (100%) and specific (88%) to detect HPS in patients with PaO2 <60mmHg and resulted in arterial blood testing in only 13% of the cohort.72 SpO2 can thus help as a screening tool for HPS and allow clinicians to select patients that require further diagnostic work-up based on the results of pulse oximetry on room air. These would include arterial blood gas analysis, echocardiography and/or nuclear medicine scanning as described below.
The usual methods to demonstrate IPVD in the clinic are contrast-enhanced echocardiography and nuclear medicine with radionuclide-labeled albumin particles. Contrast-enhanced echocardiography (transthoracic or transesophageal echocardiogram, TTE or TEE) represents the most sensitive method of detecting IPVD. It is performed by IV injection of contrast material, usually agitated saline that forms a stream of micro-bubbles 60–150μm in diameter, or indocyanine green dye. Under normal circumstances the contrast opacifies only the right heart chambers because the contrast is filtered by the pulmonary capillary bed; however, the contrast will opacify the left heart if a right-to-left cardiac shunt is present, generally appearing in the left heart within three heart beats after injection. When an intrapulmonary shunt is present, contrast generally appears in the left heart three to six heart beats after its appearance in the right heart. In the context of patient with liver disease, detection of an intrapulmonary right-to-left shunt is considered indicative of IPVDs.73 TTE is generally preferred because it is less invasive, although TEE detects IPVDs with greater specificity due to direct visualization of micro-bubbles within the pulmonary veins as they enter the left atrium, and the result may be more sensitive when performed in the upright position than in the supine position.74,75 The detection of IPVDs by this method in patients with liver disease is insufficient to diagnose HPS; impaired oxygenation is also required.
Another method for detecting IPVD is radionuclide lung perfusion scanning, using technetium-labeled macroaggregated albumin particles (MAA). This technique involves intravenously injecting albumin macroaggregates that should be trapped in the pulmonary capillary bed, since the 20–50μm diameter of the macroaggregates exceeds the normal pulmonary capillary diameter of 8–15μm. Scans that identify uptake of the radionuclide by the kidneys and/or brain suggest that macroaggregates passed through either an intrapulmonary or intracardiac shunt. The MAA scanning offers an advantage over TTE/TEE; a positive scan (shunt fraction >6%) is specific for the presence of HPS even in the context of intrinsic lung disease; however, technetium-labeled macroaggregated albumin scanning has the inability to distinguish intrapulmonary from intracardiac shunts and has lower sensitivity than contrast-enhanced TTE/TEE.76 In potential LT candidates, regardless of the presence or absence of symptoms, screening for HPS is important and cost-effective.77 There is no standard pre-transplant screening for cardiopulmonary disease, and methods and criteria used vary from center to center. Nonetheless, detecting all HPS patients by applying a diagnostic threshold of a resting PaO2<70mmHg during liver transplantation evaluation is a reasonable goal to identify patients who will qualify for liver transplant and for whom MELD exception criteria may apply.62
TreatmentThere are no effective medical therapies for HPS, although many approaches have been attempted to improve gas exchange and decrease hypoxemia. LT offers the most promise for the successful treatment of selected patients with HPS (see below).
Medical therapyInhaled NO, somatostatin, almitrine, indomethacin, aspirin, β blockers, norfloxacin, and plasma exchange have all been tried without clear benefit.62 In one study, pentoxifylline was associated with improvement in gas exchange in patients with HPS,78 but another study noted no improvement of arterial gas exchange and poor tolerability because of gastrointestinal toxicity.79
Invasive therapyAlthough improvement of HPS has been reported in a few cases where TIPS were placed, there is no clear benefit to support the role of this strategy in the routine management of patients with HPS. Furthermore, the risk of exacerbating the hyperdynamic circulatory state typical of advanced liver disease, and thereby enhancing pulmonary vasodilatation and increasing the severity of the HPS, do not provide support for its use as a palliative strategy either.80
In the case of HPS associated with angiographically visible and discrete pulmonary arteriovenous shunts, embolization by interventional radiology with closure of such communications has been shown to improve arterial oxygenation, at least temporarily in some cases.81
HPS and liver transplantationLT is currently the only proven effective therapy for HPS and should be considered when severe hypoxemia is present; it has been associated with improvement in arterial gas exchange in over 85% of patients, with subsequent resolution or improvement of hypoxemia. In the largest single-institution series, patients with HPS had a 5-year survival of 76% after LT, a rate not significantly different from that reported in LT recipients without HPS.69 The severity of preoperative hypoxemia, underlying liver disease and comorbidities appear to be factors that increase postoperative mortality.62
In the United States, UNOS allows patients with confirmed HPS be listed for LT. When PaO2 <60mmHg on room air in the sitting position, patients can receive exception MELD scores, starting at 22 points with upgrades of 2–3 MELD points allowed every 3 months while PaO2 <60mmHg and until LT is performed. Each upgrade is equivalent to a 10% mortality increase on the waiting list.82
In patients with HPS who have undergone LT, the perioperative period may present particular clinical challenges, worsening hypoxemia may occur in the early postoperative period and many patients recover slowly resulting in prolonged intensive care unit stay and unique postoperative complications. As such, these patients should be cared for in liver transplant centers with expertise in the management of HPS and its complications.
ConclusionLiver disease and portal hypertension are associated with hypoxemia through several mechanisms, with pulmonary vascular complications conferring the greatest impact on prognosis in such patients. POPH and HPS are relatively common cardiopulmonary complications, particularly if proper and adequate screening is made in LT referral centers, where the prevalence has been shown to be the highest. POPH and HPS should be sought routinely as part of the evaluation of patients being considered for LT as most cases are asymptomatic or present with nonspecific symptomatology. Understanding the hemodynamic diagnostic criteria for POPH is paramount for adequate therapeutic decision-making and to consider withdrawing LT in the most severe stages of POPH. In contrast, the more severe forms of HPS represent an absolute indication for LT, being the definitive invasive therapy for HPS. Novel PAH-specific therapies for POPH represent an evolving area of clinical research, expanding the pool of potentially transplantable patients afflicted with POPH, with the primary goal of improving patient survival.
Financial disclosuresNone.
Conflict of interestNone.
We would like to thank Vanessa Cotchery for reviewing the grammatical aspect of the manuscript.

