

Alcohol is the most accepted addictive substance worldwide and its consumption is related to multiple health, economic, and social problems. The liver is the organ in charge of ethanol metabolism and it is susceptible to alcohol's toxic effects.
ObjetivosTo provide a detailed review of the role of oxidative stress in alcoholic liver disease and the mechanisms of damage involved, along with current information on the hepatoprotective effectiveness of the molecules that have been studied.
Materials and methodsA search of the PubMed database was conducted using the following keywords oxidative stress, alcoholic liver damage, alcoholic cirrhosis, and antioxidants. There was no time limit for gathering all available information on the subject at hand.
ResultsAccording to the literature reviewed, oxidative stress plays an important role in the pathogenesis of alcoholic liver damage. Molecules such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), formed during ethanol metabolism, structurally and functionally modify organic molecules. Consequently, biologic processes are altered and hepatocytes are sensitized to the action of cytokines like tumor necrosis factor-α, as well as to the action of endotoxins, activating signaling pathways such as those controlled by nuclear factor kappa B, extracellular signal regulated kinases, and mitogen activated protein kinase.
ConclusionsOxidative stress plays an important role in the development of liver damage resulting from alcohol consumption. The molecules that have currently displayed a hepatoprotective effect in preclinical and clinical trials must be studied further so that their effectiveness can be confirmed and they can possibly be used as adjuvant treatments for this disease.
El alcohol es la sustancia adictiva más aceptada mundialmente y su consumo está relacionado con múltiples problemas de salud, económicos y sociales. El hígado es el órgano encargado del metabolismo del etanol y es susceptible de sufrir los efectos tóxicos generados por este.
ObjetivoProveer una revisión detallada del papel del estrés oxidativo en la enfermedad hepática alcohólica, los mecanismos de daño involucrados, así como información actual de moléculas cuya eficacia hepatoprotectora ha sido investigada.
Materiales y métodosSe consultó la base de datos PUBMED utilizando como palabras clave: estrés oxidativo, daño hepático por alcohol, cirrosis alcohólica y antioxidantes, sin límite de tiempo para recabar toda la información disponible acerca de este tema.
ResultadosConforme a la literatura consultada, el estrés oxidativo desempeña un papel importante en la génesis del daño hepático por alcohol. Moléculas como las especies reactivas de oxígeno (ERO) y las especies reactivas de nitrógeno (ERN), formadas durante el metabolismo del etanol, modifican estructural y funcionalmente moléculas orgánicas alterando procesos biológicos y sensibilizando a los hepatocitos a la acción de citocinas como el factor de necrosis tumoral-α, así como a la acción de endotoxinas, activando rutas de señalización como las controladas por el factor nuclear kappa-B, las cinasas reguladas por la señalización extracelular 1/2 ERK1/2 y las proteína cinasa activada por mitógenos.
ConclusionesEl estrés oxidativo tiene un papel importante en el desarrollo del daño hepático por alcohol y las moléculas que actualmente han mostrado un efecto hepatoprotector en ensayos preclínicos y clínicos necesitan someterse a más estudios que demuestren su eficacia para considerarlos como tratamientos adyuvantes de esta enfermedad.
The most socially accepted addictive substance worldwide is alcohol. The consumption of alcoholic beverages is a hallmark of social gatherings. However, in many societies the consumption of these beverages in excess represents serious health and economic problems.1,2 Despite this fact, drinking in moderation is not considered a health risk.3–6 Chronic or excessive alcohol consumption can put physical and mental health at risk, damaging different organs such as the brain, liver, heart, lungs, skeletal musculature, and bones. Its abuse is also associated with social problems, including traffic accidents, social violence, divorce, low productivity, child abuse, and other crimes.7,8
The liver is the main site of ethanol metabolism and the principal target organ of alcohol-induced damage. The susceptibility of the liver to alcohol-induced toxicity is due to the high concentrations of alcohol present in the portal blood, as well as to the metabolic consequences of ethanol metabolism. Alcoholic liver disease covers a spectrum of stages that includes steatosis (fatty liver), steatohepatitis, and in severe cases, fibrosis and/or cirrhosis.9
Hepatic fibrosis can be regarded as an integrated and highly dynamic cellular response to chronic liver damage.10 Its progression is characterized by the perpetuation of necrosis of the parenchyma, chronic hepatitis, and qualitative and quantitative alterations in the composition of the extracellular matrix (ECM), while the activation of hepatic stellate cells (HSC) and the participation of macrophages and Kupffer cells predominate at the cellular level.10–12 At the molecular level, growth factors, cytokines, chemokines, changes in ECM organization and composition, as well as oxidative stress-related molecules, are seen to play an important pathologic role.10–12 The participation of oxidative stress in almost all clinical and experimental conditions of chronic liver disease of different etiologies, including alcohol consumption, has been demonstrated.13–15
MethodsThe PubMed database was searched using the following keywords oxidative stress, liver damage from alcohol, alcoholic cirrhosis, antioxidants. There was no time limit and the words AND and OR were used as logical combining terms, enabling us to limit the information. The original and review articles resulting from the search were selected in accordance with the main aim of this study.
Free radicals, reactive oxygen species and reactive nitrogen speciesAll molecules have electrons as peripheral components and their behavior determines the molecular properties. The stability of a molecule depends on the pairing of its electrons, and so any situation in which a species with an unpaired electron is produced, could result in a potentially reactive entity known as a free radical. Free radicals can be produced in biologic systems through a variety of processes.16 The terms free radical and reactive oxygen species (ROS) are used as equivalents, but the term ROS refers to those chemically reactive molecules derived from oxygen.17 ROS include the superoxide radical, the hydroxyl radical, hydrogen peroxide, and hypochloric acid. On the other hand, the reactive nitrogen species (RNS) refer to nitric oxide and the molecules derived from it, such as peroxynitrite and nitrogen dioxide. The RNS have a longer half-life than the ROS, making them more damaging18 (Table 1).
Reactive oxygen species and reactive nitrogen species involved in alcohol-induced hepatic damage.
| Reactive oxygen species | Reactive nitrogen species |
| Superoxide radical(O2•−)Hydroxyl radical (HO•)Radical Hydroperoxide radical (HO2•)Lipid radical (L•)Peroxy lipid radical (LO2•)Hydrogen peroxide (H2O2)Lipid hydroperoxide (LOOH) | Peroxynitryl radical (ONOO•)Nitrogen dioxide (NO2)Nitric oxide (NO) |
Different types of cells produce ROS in the liver through diverse mechanisms, after ethanol consumption.19 Hepatocytes use mechanisms such as CYP2E1, mitochondrial respiratory chain alterations, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) to produce ROS.20–22 In cytosol, ethanol metabolism by dehydrogenase alcohol enables both the formation of acetaldehyde and ROS23 and the production of nicotinamide adenine dinucleotide + hydrogen (reduced form) that can interfere with electron transference in the mitochondrion, facilitating ROS production.24 In addition, acetaldehyde production increases mitochondrial damage, enabling oxygen to be reduced to superoxide.25
CYP2E1, the isoform of the cytochrome P450 family, has an especially high index of NADPH oxidase activity, allowing the production of large quantities of the superoxide radical and hydrogen peroxide. CYP2E1 content has a positive correlation with NADPH oxidase activity and lipid peroxidation in the hepatic microsomes of alcoholic subjects and rodents fed with ethanol.26 An increase in ROS production was also apparent in HepG2 cells transfected with a CYP2E1 gene.27 Thus the capacity of this isoform for reducing O2 into the superoxide radical and hydrogen peroxide can be considered a key factor contributing to oxidative stress during chronic exposure to ethanol.
Treatment with ethanol has resulted in Nox activation in hepatocytes, enabling an increase in superoxide radical production.28 Kupffer cells are without a doubt the main source of ROS, expressing the NADPH oxidase complex; this is the central mechanism for producing these molecules.28,29 In contrast, the HSC are a small source of ROS and these pro-oxidant species participate as intracellular signaling mediators.30 Another mechanism involved in the increase of ROS during ethanol consumption is related to failures in complex I and III of the mitochondrial electron transporting chain. Reduced levels of the sulfur-iron centers as a consequence of chronic alcohol consumption have been reported.31 This impedes flavin-semiquinone mononucleotide electron transference and increases the production of superoxide anion within complex I. These lesions could increase the electron transfer to the oxygen molecule thus increasing the levels of superoxide anion and hydrogen peroxide within the mitochondrion.30
Finally, the RNS are produced mainly due to inducible nitric oxide synthase action that is expressed in all hepatic cells. Nitric oxide is obtained from this reaction and when it reacts with an oxygen molecule it facilitates the formation of the peroxynitrite anion, which can finally react with certain metallic ions or favor protein S-nitrosylation.16
The noxious effect of reactive oxygen species in lipidsPolyunsaturated fats are essential for complete cell system support, including membranes, endoplasmic reticulum, and mitochondria. The alteration of their structural properties can have terrible consequences for cell function. Lipid peroxidation has customarily been the main effect that free radicals have had on the lipids that make up the cell membranes. As a result of this process many of the trial methods are based on establishing the damage induced by free radicals, measuring the products of the reactions of these molecules on the membrane lipids. Some such products are malondyaldehyde (MDA), 4-hydroxy-2,3-nonenal (4-HNE), and 4-hydroxy-2,3-alkenal, whose concentration is proportional to the oxidative damage produced.32 In 1966, Di Luzio33 was the first to observe lipid peroxidation after exposure to alcohol, and it has been confirmed in other studies.34 Lipid peroxidation produces electrophiles, such as MDA or 4-HNE, and they can modify essential proteins. This results in the loss of essential protein function and cell homeostasis. Increases in the products released through lipid peroxidation have been observed in isolated mitochondria of rats treated with ethanol.34,35
Noxious effect of reactive oxygen species in proteinsOxidative modifications in proteins can lead to alterations in their functions, as well as their structure, which in turn results in these oxidized proteins undergoing proteolysis. The oxidative damage in these molecules is carried out in 3 stages: in the first stage, a protein can be slightly modified, but its main structure is intact, resulting in a slight reduction of its activity. In the second stage, the damage inflicted upon the protein is enough to cause a partial unfolding of the protein and the hydrophobic sequences that are generally covered inside the soluble globular proteins remain exposed on the surface. In the third stage, if the damaged protein has not been identified and degraded into proteasomes, it forms an aggregate with other proteins, lipids, and sugars.36
There is little information on protein oxidation after ethanol consumption. High concentrations of carbonylated proteins in plasma and erythrocytes in patients with chronic alcoholism have recently been reported.37 Data obtained by Mutlu-Turkoglu38 concurred with these observations.
Noxious effect of reactive oxygen species in DNAROS production and degradation can be exogenous, as well as endogenous, and an imbalance in these processes can cause damage at the DNA level, which can lead to modifications of and severe consequences for the cell.39 Of the ROS, the hydroxyl radical reacts with DNA, specifically with the carbon atoms that are forming double bonds in the nitrogenous bases and abstract a hydrogen from the methyl group of thymine, as well as from each of the carbon-hydrogen bonds of sugar (2¿-deoxyribose).40 Guanine is one of the bases with the greatest propensity for oxidative damage. More than 20 oxidation products of the guanine base have been identified and among them, the most abundant and well-studied is 8-oxo-7,8-dihydro-desoxyguanine; when it is not repaired it becomes mutagenic, because it pairs with adenine instead of cytosine.41
Morphologic and functional abnormalities are one of the first manifestations of ethanol-induced damage to hepatocytes. Different hypotheses have been formulated to explain the poor functioning of this organelle. One of the first hypotheses is linked to the loss of DNA integrity and inadequate protein synthesis. Mitochondria obtained from rats treated with ethanol showed modifications in their DNA.42 Mitochondrial DNA deletions are 8 times more frequent in the livers of alcoholic patients compared with controls.43 In addition, mitochondrial protein synthesis inhibition, linked with mitochondrial DNA damage and ribosomal defects, contributes to diminishing the functionality of the oxidative phosphorylation system, resulting in an accumulation of the reduced respiratory chain carriers in the complexes I and II after chronic ethanol consumption.44–46
Oxidative hepatic damage produced by ethanol consumptionThe capacity of ethanol to increase levels of ROS and RNS, lipid peroxidation, DNA, and proteins was demonstrated in a variety of systems, cells, and species.
Initially, alcohol consumption favors an increase in intestinal permeability and the translocation of bacterial lipopolysaccharide (LPS) from the intestine to the liver. In Kupffer cells, this LPS interacts with its receptor, TLR4, to begin the signaling that favors the production of oxidative stress and proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), in turn, facilitating liver damage.47–50
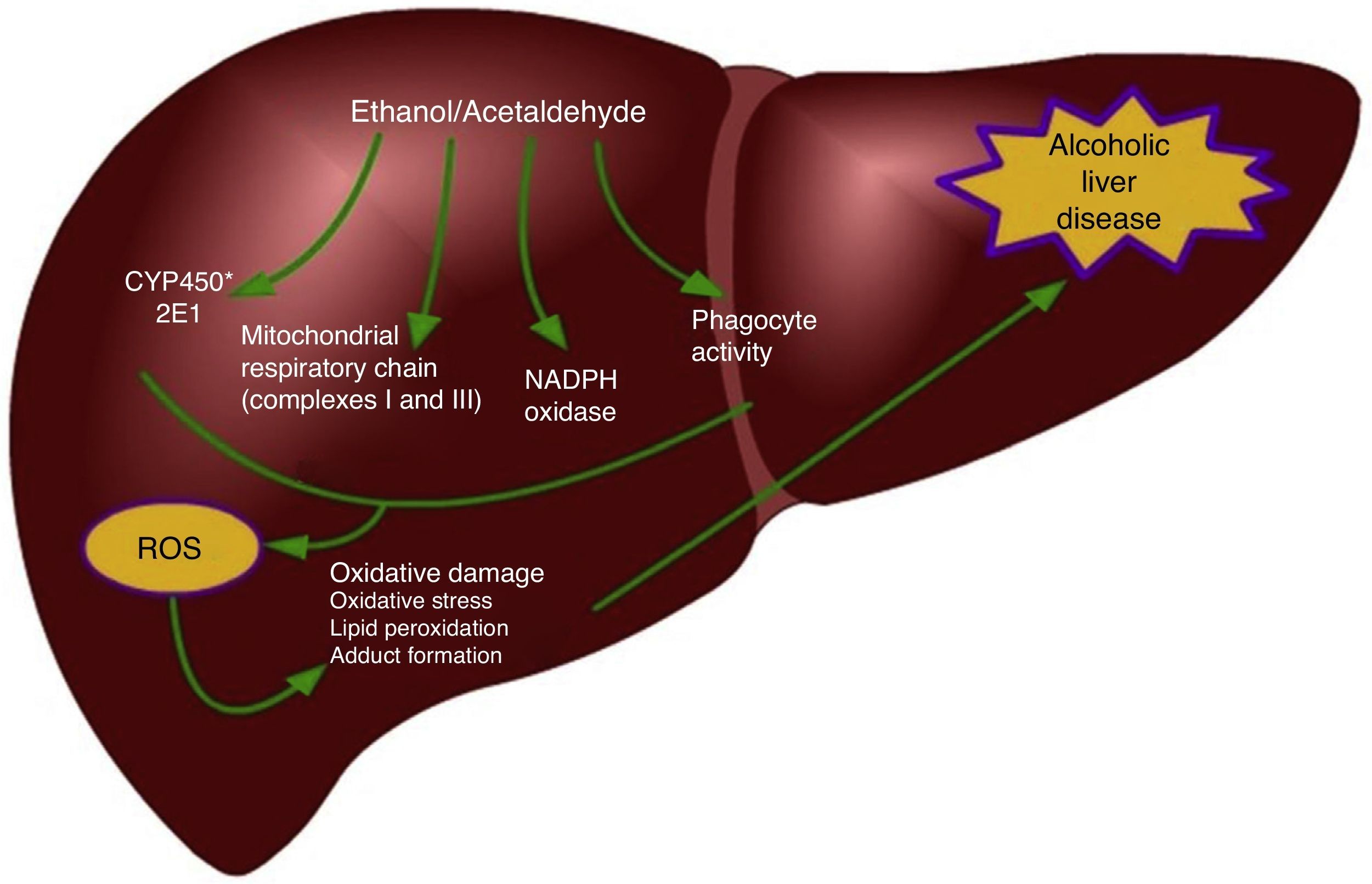
One of the main determinants of oxidative stress during alcohol consumption is the induction of the 2E1 isoform of the P450 cytochrome family into the hepatocytes and Kupffer cells,39,51 but not into the HSC.52 CYP2E1 induction is responsible for the metabolism of ethanol into acetaldehyde, but it also increases the vulnerability of alcoholics to the toxicity of different drugs, industrial solvents, and anesthetics.51 The mechanisms responsible for oxidative stress induced by ethanol consumption include intrahepatocyte formation of ROS by the mitochondrial respiratory chain, ethanol biotransformation, and the extracellular production of ROS by phagocytes (Fig. 1).53,54 The activity of CYP2E1 on ethanol, in particular, enables the formation of the hydroxyethyl free radicals (HER).53 The Kupffer cells and neutrophils infiltrated in the liver represent an important source of oxidizing species and contribute to causing oxidative stress during alcohol consumption.55 At any rate, the interaction between oxidative stress and inflammation for the progression of alcoholic liver disease is more complex. For example, oxidative damage is involved in the sensitizing of hepatocytes to the pro-apoptotic action of the TNF-α cytokine. In addition, the ROS produced by CYP2E1 and NADPH+ oxidase activity exacerbate the Kupffer cell response to endotoxins by highly regulating the Toll-like 4 (TLR-4) receptor expression.56 They also increase the transduction of signals mediated by TLR-4 through transcription factors, such as the nuclear factor kappa-B (NFκB) and STAT3. In addition, kinases activated by extracellular signals (ERK1/2) and the mitogen-activated protein kinase (MAPK) called p38, thus producing an increase in the levels of pro-inflammatory cytokines, such as TNF-α or IL-1β, or anti-inflammatory cytokines, such as IL-10 and IL-6.57–59 Recent evidence suggests that ethanol stimulates the Notch pathway that is involved in multiple inflammatory diseases such as enterocolitis, vasculitis, and bronchitis.60,61 Different experimental models indicate that there is a link between the Notch1-mediated signaling and NFκB, which is a central regulator of cellular stress in all types of cells in the liver and its activation may contribute to the development of alcoholic steatohepatitis.62,63 Wang et al. demonstrated that ethanol induces Notch1 signaling in hepatocytes that overexpressed CYP2E1, indicating that Notch1 activation is dependent on oxidative stress.64

The capacity of ethanol to start the immune response was first suggested 20 years ago, by observing antibodies directed at protein adducts with hepatic proteins.65 Oxidative stress has recently been implicated in the induced immunity in alcoholic liver disease through the detection of antibodies that selectively recognize epitopes derived from the interaction of the HERs and hepatic proteins.66 Anti-HER Ig-G recognizes CYP2E1 modified by HER as its main antigen, and the presence of anti-HER antibodies correlates with CYP2E1 activity in both rats and humans.66 However, the immune response induced by oxidative stress is not limited to the HER adducts; elevated circulating IgG titers circulating toward epitopes derived from the modification of proteins and final products of lipid peroxidation, such as MDA and 4-HNE have been observed, as well as lipid hydroperoxides that are prevalent in patients with advanced alcoholic liver disease.67 Moreover, the combined reaction of MDA and acetaldehyde with protein residuals of lysine creates condensation products called MDA-acetaldehyde adducts (MAA) that are antigenic68 and stimulate immune reactions in patients with alcoholic liver disease.69 Approximately 35% of the patients with an advanced liver disease due to alcohol also show a response to T CD4+ lymphocytes toward MDA-derived antigens, indicating that the oxidative mechanisms promote a humoral, as well as a cellular, immune response.70
Another pathway involved in hepatic damage caused by ethanol consumption and oxidative stress is one that is modulated by the adenosine-monophosphate-activated protein kinase (AMPK) in different cell types, including neurons, heart cells, skeletal muscle cells, pancreatic cells, and hepatic cells.71 AMPK has an important role in cell survival during metabolic stress, due to its ability to maintain metabolic homeostasis. It also controls the redox state and mitochondrial function. It is known that the pathways associated with AMPK can suppress oxidative stress-induced cell death.72 This protein is important in the development of ethanol-induced steatosis, since AMPK inhibition increases lipogenesis and inhibits fatty acid β-oxidation.73 The inhibition of AMPK enables acetyl-CoA carboxylase activation, improving the levels of malonyl-CoA, which inhibits fatty acid absorption and β-oxidation at the mitochondrial level. In animal models, AMPK activation by adiponectin and other agents, such as rosiglitazone, has been shown to improve ethanol-induced fatty liver, demonstrating the key role that AMPK protein inhibition has in the development of steatosis.74,75 Nevertheless, AMPK inhibition is paradoxical because ethanol metabolism is accompanied by ROS production, critical factors in helping control the AMPK activation mediated by different stimuli such as hyperglycemia and the pharmacologic treatment with metformin.76 These results suggest that AMPK inhibition due to alcohol consumption and AMPH activation by ROS occur through different mechanisms.
Moreover, the first studies demonstrating the effects of oxidative stress on AMPK activation were carried out more than 10 years ago, when Choi et al.77 showed that the AMPK cascade was highly sensitive to ROS.
The signaling pathway mediated by the nuclear factor-erythroid 2-related factor-2 (Nrf2)-ARE also plays an important role in the development of alcoholic liver disease. The induction of many cytoprotective enzymes in response to chemical stress is primarily regulated at the transcriptional level by a protein called ARE (a Cis-regulating element) that initially was found in the promoter genes that encode for 2 important detoxifying enzymes: glutathione S-transferase and NADPH:quinone oxidoreductase.78 Gene transcription activation through ARE is mediated by Nrf2. There is evidence that the levels of the protein, as well as the mRNA of Nrf2, are elevated in hepatic tissue and in the hepatocytes of animals chronically treated with ethanol. HepG2 cells overexpressed the CYP2E1 isoform and also showed an increase in mRNA and Nrf2 protein expression compared with control HepG2 cells.79 Other reports attribute an important role to Nrf2 in the protection against ethanol-induced hepatic damage. Nrf2 knockout mice that were chronically given ethanol were observed to have a significantly higher mortality rate than wild type animals;80 a diminished ability to detoxify acetaldehyde was also observed in the knock-out mice, which enabled the accumulation of toxic metabolites.80
Despite the difficulty of experimentally reproducing a state of alcoholic fibrosis in animal models, different studies have shown that lipid peroxidation precedes the initiating signals of fibrosis and is associated with an increase in profibrogenic cytokine production, such as transforming growth factor β (TGF-β).81 It has also been reported that lipid peroxidation induced by ethanol consumption promotes NFκB transactivation, thus favoring α2 (1) collagen promoter gene expression in the HSC through the stimulation of the kinase cascade, in which PKC, PI3K, and PKB/Akt participate.82 These observations are consistent with the role of 4-HNE that acts as a profibrogenic stimulus for collagen production in human HSCs.83
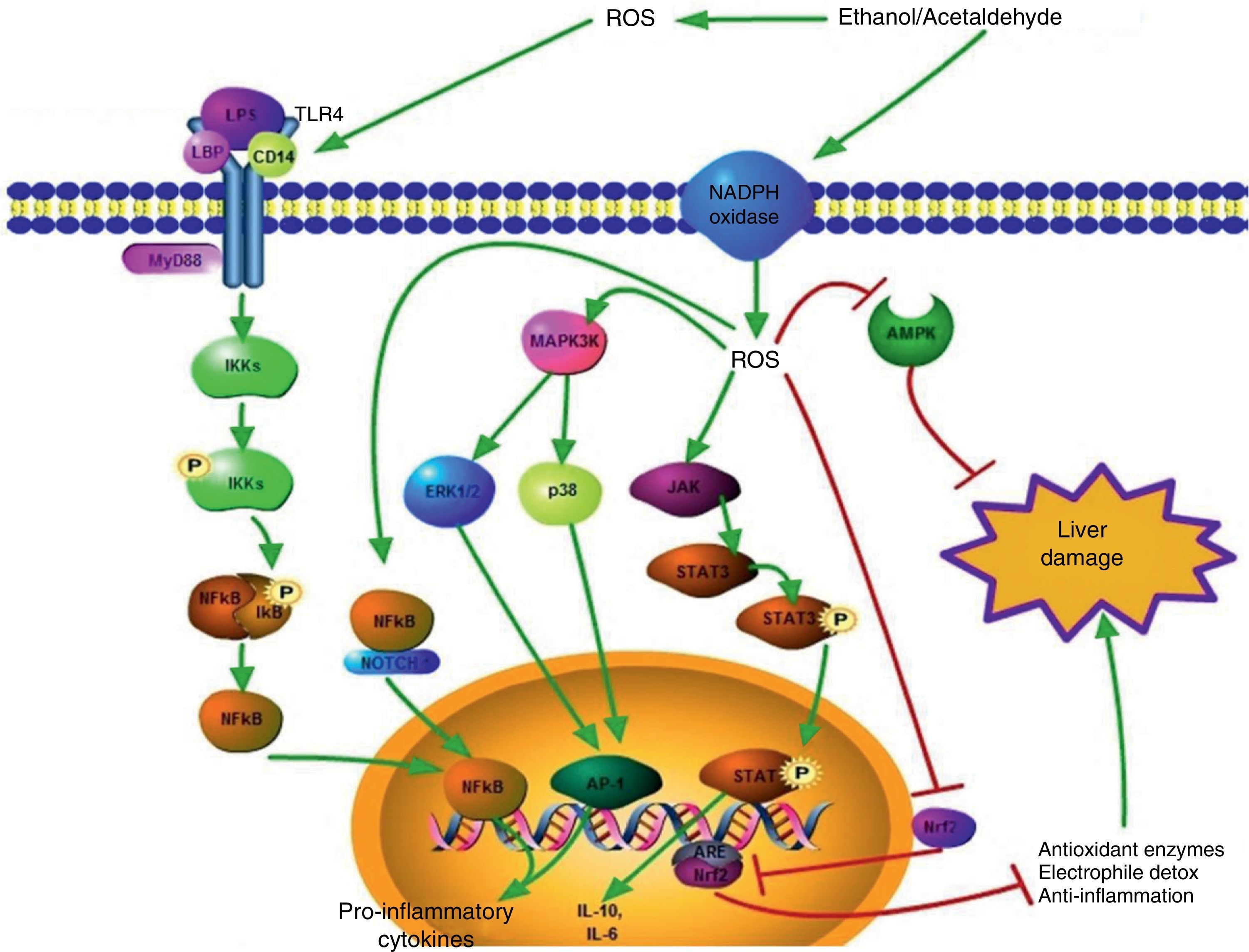
A summary of these signaling pathways is illustrated in Figure 2.

Signaling pathways activated by the ROS generated during ethanol consumption. ROS that are produced extracellularly have the capacity to promote TLR-4 overexpression, favoring the pathway of damage mediated by LPS. This leads to the translocation of the NFκB transcription factor to the nucleus and thus proinflammatory cytokine gene transcription takes place. Intracellular ROS can also favor NFκB activation through the NFκB/Notch1 complex, which increases the damage.
In regard to the cytoplasm, ROS generated from different sources, such as oxidase NADPH activity, activate different proteins related to hepatic damage such as ERK1/2, p38, and JAK, which promote the activation of transcription factors such as AP1 and STAT3, positively modulating pro and anti-inflammatory cytokine gene expression, respectively.
The cell signaling related to the hepatoprotective response is also altered by ROS; the pathways induced by AMPK and Nrf2/ARE are inhibited by the action of the oxidizing species.
The green arrows indicate response promotion and the red arrows indicate response inhibition.
AMPK: adenosine monophosphate-activated protein kinase; ARE: antioxidant response element; ERK1/2: extracellular signal-regulated kinase 1/2; JAK: Janus kinase, LPS: lipopolysaccharide; NFκB: nuclear factor κB; Nrf2: nuclear factor erythroid 2-related factor 2; ROS: reactive oxygen species.
Given that multiple factors are involved in the production of oxidative stress during alcoholic liver disease, sex and idiosyncrasy are points that should be taken into account.
Women develop a more severe form of alcoholic liver disease with less consumption and fewer years of exposure to this hepatotoxin. The exact mechanisms are unknown, but there is evidence suggesting that the sex hormones play an important part.84 Women with chronic alcoholism have menstrual cycle disorders that include amenorrhea, anovulation, or irregular cycles and luteal phase dysfunction.85,86 Studies carried out on rats have shown that alcohol ingestion conditions low levels of estrogen and anovulatory cycles characterized by a decrease in progesterone.87 Similar responses have been observed in premenopausal alcoholic women. These results suggest that estrogens (and progesterone) are important in the development of alcoholic liver disease and the hormonal impact can be different depending on the stage of life of the woman (menopause, postmenopause), with alcohol ingestion having an important role in producing damage.
On the other hand, all the phases that participate in the development of alcoholic liver disease are regulated by genes, that either by themselves or in combination, represent risks and influence the reactions with ethanol.88 After its absorption, ethanol is degraded in the liver and other tissues by the alcohol dehydrogenase (ADH) enzyme in the cytosol and by CYP2E1 in the microsomes. There are different kinds of ADH and 2 of the known human ADH genes are the polymorphic ADH2 and ADH3, with 3 existing alleles each, and they reveal important enzymatic characteristics. The alleles for ADH2 are ADH2*1, found in Caucasians, and ADH2*2, found in Asians. According to the differences in the capacity for metabolizing alcohol into acetaldehyde, it has been speculated that individuals with the ADH2*2 allele are at greater risk for developing hepatic damage due to higher acetaldehyde exposure.89 There is a similar situation with the genetic variants of CYP2E1; the mutant CYP2E1 c2 allele is associated with greater gene transcription, protein level, and enzyme activity than the c1 allele, which can result in greater exposure of the liver to acetaldehyde and the ROS, exacerbating hepatic damage.88
Antioxidant defenses in alcoholic liver diseaseDuring ethanol consumption different pathways are activated that lead to ROS overproduction. However, such massive production often surpasses the capacity of the endogenic antioxidant defenses to counteract the generated damage and this phenomenon is known as oxidative stress.
The organism has multiple mechanisms for confronting this phenomenon. The antioxidant defense system is made up of a group of substances that delay or prevent oxidation of an oxidizable substrate, such as carbohydrates, proteins, lipids, and DNA molecules.90
It is known that the administration of antioxidant agents, or iron, zinc, and copper chelating agents, as well as agents that replace reduced glutathione (GSH), such as N-acetyl cysteine, can attenuate or prevent the toxic effects of alcohol.91 Polyunsaturated lipids (necessary in order for lipid peroxidation to take place) replaced by medium-chain triglycerides or saturated lipids, administered intragastrically in the diet of rats, prevents or reduces lipid peroxidation and the hepatic damage induced by alcohol consumption.92,93 Interestingly, the administration of antioxidants such as ebselen, vitamin E, superoxide dismutase, and GSH precursors prevents alcohol-induced hepatic damage in rats.94
Other compounds that have shown beneficial responses to ethanol consumption-induced hepatic damage are the corticosteroids. This group of drugs can attenuate the inflammatory response during alcoholic hepatitis, reduce cytokine production, suppress acetaldehyde adduct formation, and inhibit collagen production.95 Patients with severe alcoholic liver disease that have been treated with steroids have been observed to show short-term histologic improvement; this improvement is associated with a decrease in TNF-α circulation and in intercellular-1 adhesion molecule expression, as well as with changes in the soluble intercellular-1 adhesion molecule in the hepatic vein.96
Without a doubt, the plant that has been studied the most in relation to the treatment of liver diseases is Silybum marianum. It is made up of 4 isomers: silibinin, isosilibinin, silidianin, and silicristin. Of the 4, silibinin is the most active compound. According to the results of different experimental studies on hepatic damage, silymarin offers adequate protection exerting good antioxidant effects97 and antifibrotic effects,98 in addition to being an excellent immunomodulator and cell membrane stabilizer, as well as aiding in the process of regeneration.98–101 Clinically, silymarin has demonstrated positive effects on alcoholic liver disease, hepatic cirrhosis, drug-induced hepatic damage, and in diabetic patients.101
Metadoxine (pyridoxol L-2-pyrrolidone-5-carboxylate), the ionic pair between pyrrolidone carboxylate and pyridoxine, is another of the compounds that has demonstrated great efficacy in the treatment of acute alcohol intoxication. Pyrrolidone carboxylate participates in amino acid metabolism through the glutathione pathway.102 It participates in de novo ATP synthesis103 and prevents the reduction of ATP levels in the brain and liver of rats with acute ethanol intoxication.104 Importantly, it is known that pyridoxine increases the metabolic degradation rate of ethanol, as well as the cell function damage caused by acetaldehyde.105 Metadoxine appears to be capable of accelerating ethanol metabolism in both rats and humans through facilitating increased activity of the dehydrogenase acetaldehyde enzyme, increasing the plasma clearing of ethanol and acetaldehyde, and the renal elimination of ketones that are a product of its metabolism.106–108 This compound has been capable of preventing GSH depletion, oxidative damage at the lipid level, the accumulation of collagen, and alcohol and acetaldehyde-induced TNF-α secretion in hepatocytes and stellate cells.109
Furthermore, metadoxine has shown beneficial effects in relation to early mortality in patients with alcoholic hepatitis at the Hospital General de México (Higuera de la Tijera, unpublished data).
It is our opinion that the abovementioned compounds are the outstanding elements in the treatment of alcoholic liver and the efficacy of many of them has been demonstrated in animal models. Such results encourage the further study of these molecules, but through controlled clinical studies, so that in the future they may be considered a tool in the pharmacologic treatment of this disease.
ConclusionsChronic ethanol consumption plays a relevant role in the generation of hepatic disease. Considering that normal ROS biosynthesis is a natural part of biochemical processes, ethanol ingestion is thought to favor oxidative stress and the promotion of inflammatory and fibrotic damage. Because there are multiple mechanisms through which alcohol facilitates hepatic damage, the development of therapeutic strategies for treating this disease is very complex, which explains the continuing lack of effective treatment for resolving this health problem. However, we believe that by blocking one of the pathways of damage through the use of antioxidants, supplements favoring de novo GSH generation, polyphenolic agents, such as vitamin E or silymarin, and drugs, such as metadoxine, could improve the quality of life of these patients.
Financial disclosureMarina Galicia Moreno received financial support through the postdoctoral Scholarship Program of the UNAM, DGAPA 2013-2014, and the PAPIIT IA203113 program.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Galicia-Moreno M, Gutiérrez-Reyes G. Papel del estrés oxidativo en el desarrollo de la enfermedad hepática alcohólica. Revista de Gastroenterología de México. 2014;79:135–144.

