

El alcohol es la sustancia adictiva más aceptada mundialmente y su consumo está relacionado con múltiples problemas de salud, económicos y sociales. El hígado es el órgano encargado del metabolismo del etanol y es susceptible de sufrir los efectos tóxicos generados por este.
ObjetivoProveer una revisión detallada del papel del estrés oxidativo en la enfermedad hepática alcohólica, los mecanismos de daño involucrados, así como información actual de moléculas cuya eficacia hepatoprotectora ha sido investigada.
Materiales y métodosSe consultó la base de datos PUBMED utilizando como palabras clave: estrés oxidativo, daño hepático por alcohol, cirrosis alcohólica y antioxidantes, sin límite de tiempo para recabar toda la información disponible acerca de este tema
ResultadosConforme a la literatura consultada, el estrés oxidativo desempeña un papel importante en la génesis del daño hepático por alcohol. Moléculas como las especies reactivas de oxígeno (ERO) y las especies reactivas de nitrógeno (ERN), formadas durante el metabolismo del etanol, modifican estructural y funcionalmente moléculas orgánicas alterando procesos biológicos y sensibilizando a los hepatocitos a la acción de citocinas como el factor de necrosis tumoral-α, así como a la acción de endotoxinas, activando rutas de señalización como las controladas por el factor nuclear kappa-B, las cinasas reguladas por la señalización extracelular 1/2 ERK1/2 y las proteína cinasa activada por mitógenos.
ConclusionesEl estrés oxidativo tiene un papel importante en el desarrollo del daño hepático por alcohol y las moléculas que actualmente han mostrado un efecto hepatoprotector en ensayos preclínicos y clínicos necesitan someterse a más estudios que demuestren su eficacia para considerarlos como tratamientos adyuvantes de esta enfermedad.
Alcohol is the most accepted addictive substance worldwide and its consumption is related to multiple health, economic, and social problems. The liver is the organ in charge of ethanol metabolism and it is susceptible to alcohol's toxic effects.
ObjetivosTo provide a detailed review of the role of oxidative stress in alcoholic liver disease and the mechanisms of damage involved, along with current information on the hepatoprotective effectiveness of the molecules that have been studied.
Materials and methodsA search of the PubMed database was conducted using the following keywords oxidative stress, alcoholic liver damage, alcoholic cirrhosis, and antioxidants. There was no time limit for gathering all available information on the subject at hand.
ResultsAccording to the literature reviewed, oxidative stress plays an important role in the pathogenesis of alcoholic liver damage. Molecules such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), formed during ethanol metabolism, structurally and functionally modify organic molecules. Consequently, biologic processes are altered and hepatocytes are sensitized to the action of cytokines like tumor necrosis factor-α, as well as to the action of endotoxins, activating signaling pathways such as those controlled by nuclear factor kappa B, extracellular signal regulated kinases, and mitogen activated protein kinase.
ConclusionsOxidative stress plays an important role in the development of liver damage resulting from alcohol consumption. The molecules that have currently displayed a hepatoprotective effect in preclinical and clinical trials must be studied further so that their effectiveness can be confirmed and they can possibly be used as adjuvant treatments for this disease.
La sustancia adictiva socialmente más aceptada en el mundo es el alcohol. El consumo de bebidas alcohólicas es un sello característico de las reuniones sociales. Sin embargo, en muchas sociedades se presentan serios problemas de salud y económicos como resultado de un consumo excesivo de estas bebidas1,2, y aún así se piensa que beber con moderación no es riesgo para la salud3–6. Un consumo crónico de alcohol o un consumo excesivo pueden poner en riesgo la salud física y mental, y causar daños perjudiciales a diferentes órganos incluyendo el cerebro, el hígado, el corazón, los pulmones, el músculo esquelético y los huesos. El abuso de esta sustancia también está asociado a problemas sociales, como accidentes de tránsito, violencia social, divorcios, baja productividad, abuso de menores y otros crímenes7,8.
El hígado es el sitio principal del metabolismo del etanol y el principal órgano blanco del daño inducido por esta sustancia. La susceptibilidad del hígado a la toxicidad inducida por el alcohol es debida tanto a las altas concentraciones de alcohol presentes en sangre portal, así como a las consecuencias metabólicas del metabolismo del etanol. La enfermedad hepática alcohólica es un espectro de estadios que incluye esteatosis (hígado graso), esteatohepatitis y, en casos severos, fibrosis y/o cirrosis9.
La fibrosis hepática puede ser considerada como una respuesta celular integrada y altamente dinámica del daño hepático crónico10. La evolución de esta se caracteriza por la perpetuación de la necrosis del parénquima, hepatitis crónica y alteraciones tanto cualitativas como cuantitativas en la composición de la matriz extracelular (MEC), mientras que la activación de las células estelares hepáticas (por sus siglas en inglés HSC) y la participación de macrófagos y las células de Kupffer predominan a nivel celular10–12. A nivel molecular, factores de crecimiento, citocinas, quimiocinas, cambios en la organización y composición de la MEC, así como moléculas relacionadas con el estrés oxidativo, se indica que desempeñan un papel patológico importante10–12. Existen evidencias que nos muestran la participación del estrés oxidativo en casi todas las condiciones clínicas y experimentales de la enfermedad hepática crónica de diversas etiologías, incluido el consumo de alcohol13–15.
MétodosSe consultó la base de datos PUBMED, haciendo uso de las siguientes palabras clave: estrés oxidativo, daño hepático por alcohol, cirrosis alcohólica, antioxidantes y sin límite de tiempo, además se utilizaron como combinatorios lógicos las letras Y y O, lo que nos permitió restringir la información. Los artículos tanto de revisión como originales resultantes de nuestra búsqueda fueron seleccionados de acuerdo con el objetivo propuesto en este estudio.
Radicales libres, especies reactivas de oxígeno y de nitrógenoTodas las moléculas tienen electrones como componentes periféricos y el comportamiento de estos determina las propiedades de las moléculas. La estabilidad de una molécula depende del apareamiento de sus electrones, por lo tanto, cualquier situación en la cual una especie sea generada con un par electrónico desapareado podría resultar en una entidad potencialmente reactiva denominada radical libre. Los radicales libres pueden ser producidos en los sistemas biológicos a través de una variedad de procesos16. El término radical libre y especies reactivas de oxígeno (ERO) son utilizados a la par; sin embargo. el término ERO se refiere a aquellas moléculas químicas reactivas que son derivadas del oxígeno17. Las ERO incluyen al radical superóxido, el radical hidroxilo, el peróxido de hidrógeno y el ácido hipocloroso. Por otro lado, las especies reactivas de nitrógeno (ERN) se refieren al óxido nítrico y a las moléculas derivadas de él, como lo son el peroxinitrito y el dióxido de nitrógeno. A diferencia de las ERO, las ERN tienen un tiempo de vida media más largo lo que las hace más dañinas18 (tabla 1).
Especies reactivas de oxígeno y nitrógeno implicadas en el daño hepático por alcohol
| Especies reactivas de oxígeno | Especies reactivas de nitrógeno |
| Radical superóxido (O2•−)Radical hidróxilo (HO•)Radical hidroperóxido (HO2•)Radical lipídico (L•)Radical peroxilipídico (LO2•)Peróxido de hidrógeno (H2O2)Hidroperóxido lipídico (LOOH) | Radical peroxinitrito (ONOO•)Dióxido de nitrógeno (NO2)Óxido nítrico (NO) |
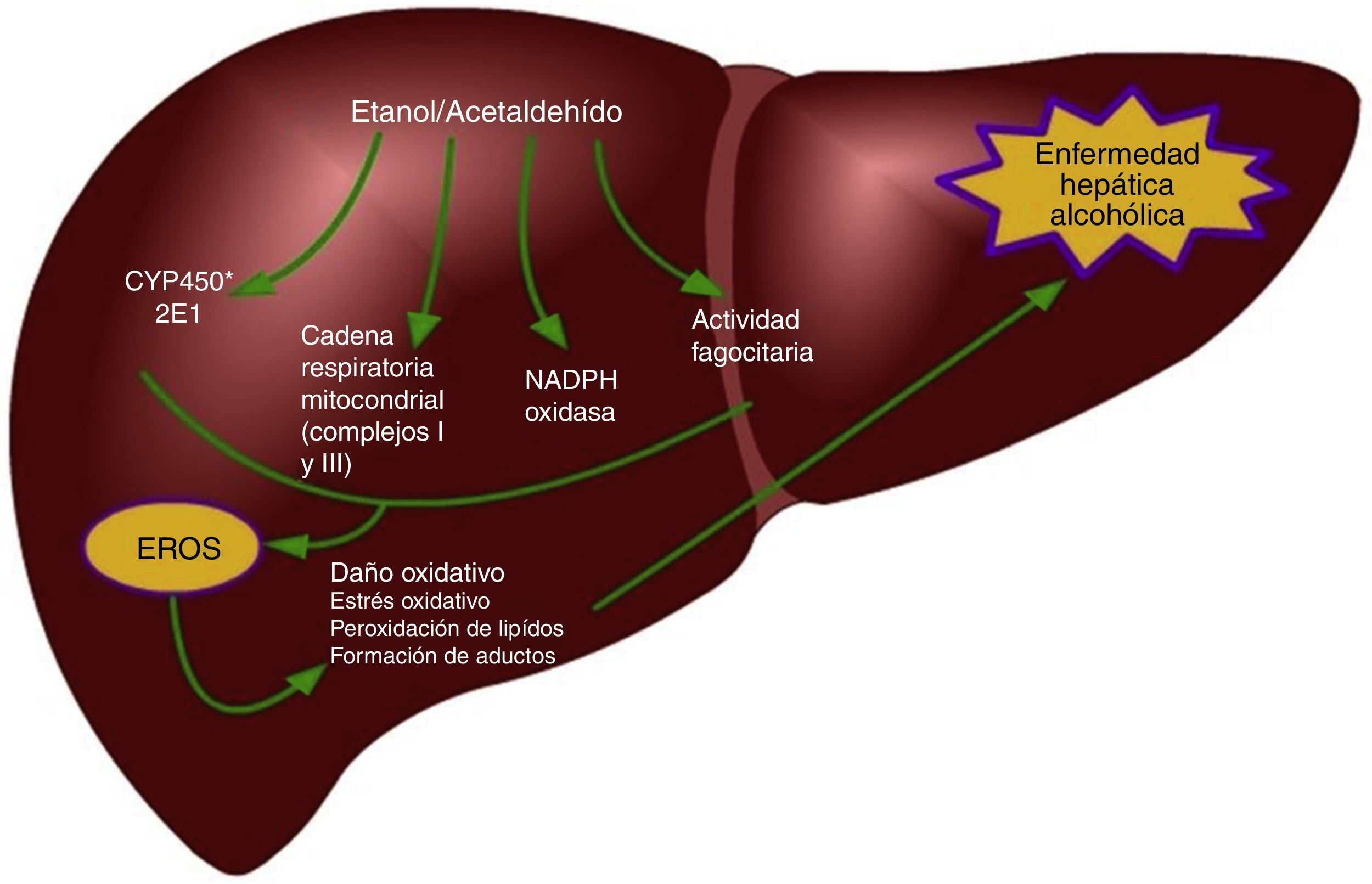
En el hígado, diversos tipos celulares generan ERO a través de diferentes mecanismos después del consumo de etanol19, los hepatocitos usan mecanismos para producir ERO como el CYP2E1, alteraciones en la cadena respiratoria mitocondrial y la nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa (Nox)20-22. En el citosol, el metabolismo del etanol por la alcohol deshidrogenasa permite la formación de acetaldehído y ROS23; además, permite la generación de la forma reducida de la nicotinamida adenina dinucleótido (forma reducida), que puede interferir con la transferencia de electrones en la mitocondria facilitando la generación de ROS24, además la formación de acetaldehído incrementa el daño mitocondrial permitiendo un incremento en la reducción de oxígeno a superóxido25.
La isoforma de la familia del citocromo P450, el CYP2E1, tiene un especial alto índice en la actividad de la NADPH oxidasa; esto permite la producción de grandes cantidades del radical superóxido y de peróxido de hidrógeno. En los microsomas hepáticos de sujetos alcohólicos y de roedores alimentados con etanol, el contenido de CYP2E1 correlaciona positivamente con la actividad de la NADPH oxidasa y la peroxidación lipídica26. Un incremento en la producción de ROS también fue evidente en células HepG2 transfectadas con un gen de CYP2E127; así, la capacidad de esta isoforma para reducir O2 en radical superóxido y peróxido de hidrógeno puede considerarse como un factor clave que contribuye al estrés oxidativo durante una exposición crónica a etanol.
Por último, se ha observado que el tratamiento con etanol también resulta en la activación de Nox en los hepatocitos, permitiendo un incremento en la producción de radical superóxido28. Las células de Kupffer son, sin duda, la principal fuente de ERO; en ellas se expresa el complejo NADPH oxidasa y este es el principal mecanismo para la producción de estas moléculas28,29; por el contrario, las HSC son una fuente minoritaria de ERO y estas especies prooxidantes participan como mediadoras de la señalización intracelular30. Otro mecanismo implicado en el incremento de las ERO durante el consumo de etanol está relacionado con fallas en los complejos i y iii de la cadena transportadora de electrones mitocondrial; se ha reportado que niveles de los centros sulfuro-hierro están disminuidos como consecuencia de un consumo crónico de alcohol31. Esto impide la transferencia de electrones del mononucleótido de flavina-semiquinona e incrementar la generación de anión superóxido dentro del complejo i; estas lesiones podrían incrementar la transferencia de electrones al oxígeno molecular y así los niveles del anión superóxido y del peróxido de hidrógeno dentro de la mitocondria estarían elevados30.
Finalmente, las ERN son producidas principalmente gracias a la acción de la sintasa de óxido nítrico inducible, que es expresada en todas las células hepáticas; de esta reacción se obtiene el óxido nítrico, el cual, al reaccionar con una molécula de oxígeno, favorece la formación del anión peroxinitrito y este finalmente puede reaccionar con algunos iones metálicos o favorecer la S-nitrosilación de proteínas16.
Efecto nocivo de las especies reactivas de oxígeno en lípidosLos lípidos poliinsaturados son esenciales para el completo soporte del sistema de la célula, incluyendo membranas, retículo endoplásmico y mitocondrias. La alteración de sus propiedades estructurales puede tener terribles consecuencias para la función celular. La peroxidación de lípidos ha sido tradicionalmente el principal efecto de los radicales libres sobre los lípidos que constituyen las membranas celulares. Como resultado de este proceso, muchos de los métodos de ensayo se basan en establecer el daño inducido por los radicales libres midiendo los productos de reacción de estas moléculas sobre los lípidos de membrana, como lo son el malondialdehído (MDA), el 4-hidroxi-2,3-nonenal (4-HNE) y el 4-hidroxi-2,3-alquenal, en donde la concentración de estos productos de reacción es proporcional al daño oxidativo generado32. Di Luzio33, en 1966, fue el primero en observar la peroxidación lipídica después de la exposición a alcohol y esto ha sido confirmado en otros estudios34. La peroxidación de lípidos produce electrófilos como el MDA o el 4-HNE; estos pueden modificar proteínas esenciales, lo que trae como resultado la pérdida de la función de esta proteína y la homeostasis celular. Se han observado incrementos en los productos liberados de la peroxidación lipídica en las mitocondrias aisladas de ratas tratadas con etanol34,35.
Efecto nocivo de las especies reactivas de oxígeno en las proteínasModificaciones oxidativas en las proteínas pueden dar lugar a alteraciones en sus funciones, así como en su estructura. Todo esto conlleva a que estas proteínas oxidadas se sometan a proteólisis. El daño oxidativo en estas moléculas se lleva a cabo en 3 etapas: en la primera etapa, una proteína puede estar ligeramente modificada, pero la estructura principal sigue intacta, lo que conlleva a una ligera disminución de su actividad; en la segunda etapa, el daño inflingido en la proteína es suficiente para causar un despliegue parcial de la proteína, también las secuencias hidrofóbicas, que generalmente se encuentran cubiertas dentro de proteínas solubles globulares, quedan expuestas en la superficie. Por último, en la tercera etapa, si la proteína dañada no ha sido identificada y degradada en proteosomas, forma un agregado con otras proteínas, lípidos y azúcares36.
Existe poca información acerca de la oxidación de proteínas después de un consumo de etanol. Recientemente, se han reportado altas concentraciones de proteínas carboniladas en plasma y eritrocitos en pacientes con alcoholismo crónico37. Datos obtenidos por Mutlu-Turkoglu et al.38 también fueron acordes con estas observaciones.
Efecto nocivo de las especies reactivas de oxígeno en el ADNLa producción y la degradación de las ERO pueden darse tanto exógena como endógenamente y un desequilibrio de estos procesos puede generar un daño a nivel de ADN, lo que puede dar lugar a modificaciones y consecuencias graves para la célula39. De las ERO, el radical hidroxilo reacciona con el ADN, específicamente con los átomos de carbono que están formando dobles enlaces en las bases nitrogenadas y abstraen un hidrógeno del grupo metilo de la timina, así como de cada uno de los enlaces carbono hidrógeno del azúcar (2¿-deoxirribosa)40. Unas de las bases más propensas al daño oxidativo es la guanina. Más de 20 productos de oxidación de la base de guanina han sido identificados y entre ellos el más abundante y bien estudiado es el 8-oxo-7,8-dihidro-desoxiguanina, el cual, cuando no es reparado, resulta ser mutagénico, ya que se aparea con la adenina en lugar de la citocina41.
Anormalidades morfológicas y funcionales de la mitocondria representan una de las primeras manifestaciones del daño a los hepatocitos inducidos por el etanol. Diferentes hipótesis han sido formuladas para explicar el mal funcionamiento de este organelo; una de las primeras hipótesis está vinculada a la pérdida de la integridad del ADN y una inadecuada síntesis de proteínas; además, las mitocondrias obtenidas de ratas tratadas con etanol mostraron modificaciones en su ADN42. Deleciones del ADN mitocondrial son 8 veces más frecuentes en hígados de pacientes alcohólicos comparados con los controles43; además, la inhibición de la síntesis de proteínas mitocondriales, vinculada con el daño del ADN mitocondrial y defectos en los ribosomas, contribuye a disminuir la funcionalidad del sistema de fosforilación oxidativa, lo que resulta en una acumulación de los acarreadores respiratorios reducidos en los complejos i y ii después de un consumo crónico de etanol44-46.
Daño hepático oxidativo generado por el consumo de etanolLa capacidad que tiene el etanol de incrementar los niveles de ERO y ERN, así como la peroxidación de lípidos, ADN y proteínas, fue demostrada en una variedad de sistemas, células y especies.
En un inicio, el consumo de alcohol favorece un incremento de la permeabilidad intestinal y la translocación del lipopolisacárido (LPS) bacteriano del intestino al hígado. En las células de Kupffer, este LPS interactúa con su receptor, el TLR4, para comenzar la señalización que favorece la generación de estrés oxidativo y la producción de citocinas proinflamatorias, como el factor de necrosis tumoral-α (TNF-α por sus siglas en inglés), lo que favorece la generación del daño hepático47-50.
Una de las determinantes principales del estrés oxidativo durante el consumo de etanol está representada por la inducción de la isoforma 2E1 de la familia del citocromo P450 en los hepatocitos y en las células de Kupffer39,51 pero no en las HSC52. La inducción del CYP2E1 es responsable del metabolismo del etanol a acetaldehído, pero también incrementa la vulnerabilidad de los alcohólicos a la toxicidad de diversos fármacos, solventes industriales y anestésicos51. Los mecanismos responsables del estrés oxidativo inducido por el consumo de etanol incluyen la formación intrahepatocito de las ERO por la cadena respiratoria mitocondrial y la biotransformación del etanol, así también por la generación extracelular de ERO por los fagocitos (fig. 1)53,54. En particular, la actividad del CYP2E1 sobre el etanol permite la formación de radicales libres hidroxietil (HER)53. Las células de Kupffer y los neutrófilos infiltrados en el hígado representan una fuente importante de especies oxidantes y contribuyen a causar un daño oxidativo durante el consumo de alcohol55. Como sea, la interacción entre el estrés oxidativo y la inflamación para la progresión de la enfermedad hepática alcohólica es más compleja. Por mencionar, el daño oxidativo está implicado en la sensibilización de los hepatocitos a la acción proapoptótica de la citocina TNF-α. Además, las ERO producidas por la actividad del CYP2E1 y la NADPH+ oxidasa exacerban la respuesta de las células de Kupffer a endotoxinas al regular a la alza la expresión del receptor Toll-like 4 (TLR-4)56 y también aumentan la transducción de señales mediada por el TLR-4 a través de factores de transcripción, como el factor nuclear kappa-B (NF-κB) y STAT3, y además, cinasas activadas por señales extracelulares (ERK1/2) y la proteína cinasa activada por mitógenos (MAPK) llamada p38, generando así un incremento en los niveles de citocinas proinflamatorias como el TNF-α o la IL-1β o citocinas antiinflamatorias, como la IL-10 e IL-657-59. Recientes evidencias indican que el etanol estimula la vía de Notch, que está implicada en múltiples enfermedades inflamatorias, como enterocolitis, vasculitis y bronquitis60,61. Diversos modelos experimentales indican que hay un vínculo entre la señalización mediada por Notch1 y el NFκB, que es un regulador central del estrés celular en todos los tipos de células en el hígado, y su activación puede contribuir al desarrollo de la esteatohepatitis alcohólica62,63. Wang et al. demostraron que el etanol induce la señalización de Notch1 en hepatocitos que sobre expresaban CYP2E1, lo cual indica que la activación de Notch1es dependiente del estrés oxidativo64.

La capacidad del etanol para dar inicio a la respuesta inmunitaria fue indicada por primera vez hace 20 años al hacer la observación de anticuerpos dirigidos a aductos de proteínas con proteínas hepáticas65. Recientemente, se ha implicado al estrés oxidativo en la inmunidad inducida en la enfermedad hepática alcohólica por la detección de anticuerpos que selectivamente reconocen epítopes derivados de la interacción de los HER y proteínas hepáticas66. Anti-HER Ig-G reconoce a CYP2E1 modificado por HER como su principal antígeno y, tanto en rata como en humano, la presencia de anticuerpos anti-HER correlaciona con la actividad del CYP2E166. Sin embargo, la respuesta inmunitaria inducida por el estrés oxidativo no está limitada a los aductos de HER; se han observados títulos elevados de IgG circulantes hacia epítopes derivados de la modificación de proteínas y productos finales de la peroxidación lipídica, como el MDA y 4-HNE, así como los hidroperóxidos lipídicos, que prevalecen en pacientes con una avanzada enfermedad hepática alcohólica67. Además, la reacción combinada de MDA y acetaldehído con residuos proteicos de lisina genera productos de condensación llamados aductos de MDA-acetaldehído (MAA), que son antigénicos68 y estimulan reacciones inmunitarias en pacientes con enfermedad hepática alcohólica69. Aproximadamente el 35% de los pacientes con una enfermedad hepática por alcohol avanzada también muestran respuesta a linfocitos T CD4+ hacia antígenos derivados de MDA, indicando que los mecanismos oxidativos promueven la respuesta inmunitaria tanto humoral como celular70.
Otra vía involucrada en el daño hepático provocado por el consumo de etanol y el estrés oxidativo es la modulada por la proteína cinasa activada por adenosil-monofosfato (AMPK) en distintos tipos celulares, incluyendo neuronas, corazón, músculo esquelético, músculo liso vascular, páncreas y células hepáticas71. La AMPK desempeña un papel importante en la sobrevivencia celular durante el estrés metabólico por su habilidad de mantener la homeostasis metabólica. También controla el estado redox y la función mitocondrial; se sabe que las vías asociadas a AMPK pueden suprimir la muerte celular inducida por el estrés oxidativo72. Esta proteína es importante en el desarrollo de esteatosis inducida por el consumo de etanol, ya que la inhibición de AMPK incrementa la lipogénesis e inhibe la β-oxidación de los ácidos grasos73. La inhibición de la AMPK permite la activación de la acetil-CoA carboxilasa, mejorando los niveles de malonil CoA, lo cual inhibe la absorción y la β-oxidación de los ácidos grasos a nivel mitocondrial. Adicionalmente, se ha demostrado que la activación de AMPK por la adiponectina y otros agentes, como la rosiglitazona, mejora el hígado graso inducido por el consumo de etanol en modelos animales, lo que demuestra el papel clave de la inhibición de la proteína AMPK en el desarrollo de la esteatosis74,75. Sin embargo, la inhibición de la AMPK es paradójica, ya que el metabolismo del etanol está acompañado de la producción de ERO y ellos son factores críticos que ayudan a controlar la activación de AMPK, que es mediada por diferentes estímulos, como la hiperglucemia y el tratamiento farmacológico con metformina76. Estos resultados indican que la inhibición del AMPK por consumo de alcohol y la activación de la AMPK por las ERO ocurre por mecanismos diferentes.
Por otro lado, los primeros estudios en los que se evidenciaban los efectos del estrés oxidativo en la activación de la AMPK surgieron hace más de 10 años cuando Choi et al.77 demostraron que la cascada de la AMPK era altamente sensible a las ERO.
La vía de señalización mediada por el factor nuclear E2 relacionado al factor 2 (Nrf2)-ARE también desempeña un papel importante en el desarrollo de la enfermedad hepática por alcohol. La inducción de muchas enzimas citoprotectoras en respuesta al estrés químico es regulada primariamente a nivel transcripcional por una proteína denominado ARE (elemento Cis-regulador), que inicialmente se encontró en el promotor de genes que codifican para 2 enzimas detoxificadoras importantes, la glutatión-S-transferasa y la NADPH:quinona óxido reductasa78. La activación de la transcripción génica a través de ARE es mediada por el Nrf2. Existen evidencias que muestran que los niveles de la proteína, así como del ARNm de Nrf2, se encuentran elevados en tejido hepático y en los hepatocitos de animales tratados de manera crónica con etanol. Células HepG2 sobreexpresaron la isoforma CYP2E1 y, de igual manera, mostraron un incremento en la expresión del ARNm y proteína de Nrf2 comparadas con células HepG2 controles79. Otros reportes adjudican un papel importante del Nrf2 en la protección contra el daño hepático inducido por etanol. En ratones knockout de Nrf2 administrados de manera crónica con etanol se observó un incremento significativo en la mortalidad de los animales comparado con los animales wild-type80; también se observó una disminución en la habilidad para detoxificar el acetaldehído en los ratones knockout, lo que permitió la acumulación de los metabolitos tóxicos80.
Si bien es difícil reproducir experimentalmente un estadio de fibrosis alcohólica en modelos animales, diversos estudios han demostrado que la peroxidación lipídica precede a las señales de inicio de la fibrosis y está asociada a un incremento en la producción de citocinas profibrogénicas como el factor de crecimiento transformante β (TGF-β)81; además se ha reportado que la peroxidación lipídica inducida por el consumo de etanol promueve la transactivación del NF-κB; de esta manera, se favorece la expresión del gen promotor de colágeno α2 (1) en las HSC a través de la estimulación de la cascada de cinasas en donde participan la PKC, PI3K y la PKB/Akt82. Estas observaciones son consistentes con el papel del 4-HNE, que actúa como un estímulo profibrogénico para la producción de colágeno en las HSC humanas83.
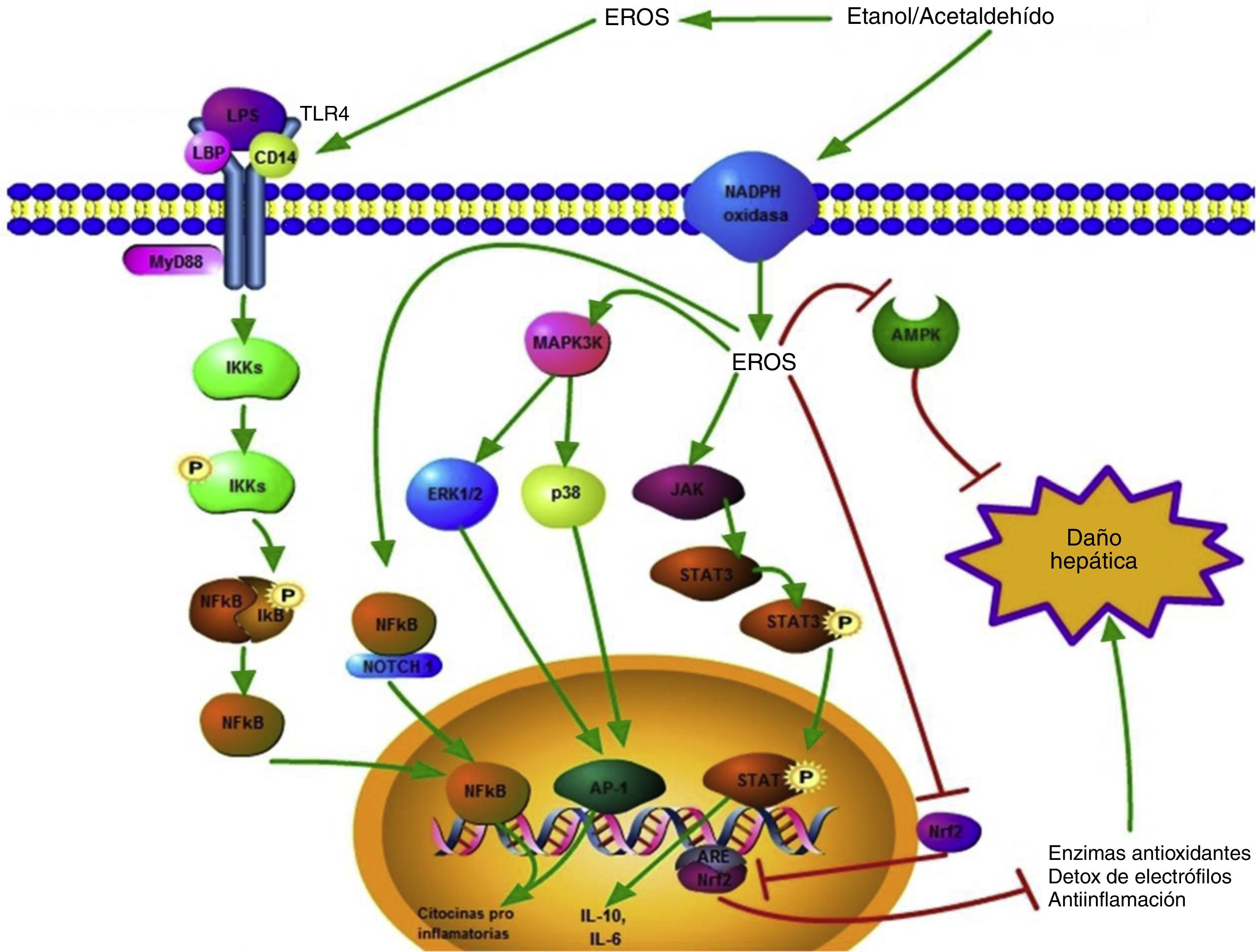
El resumen de estas vías de señalización se encuentra ilustrado en la figura 2.

Vías de señalización activadas por las ERO generadas durante el consumo de etanol. Las ERO producidas extracelularmente tienen la capacidad de promover la sobre expresión del TLR-4 favoreciendo la vía de daño mediada por el LPS; esto conlleva a la translocación del factor de transcripción NF-κB al núcleo y así la transcripción génica de citocinas proinflamatorias se lleva a cabo. Las ERO intracelulares también pueden favorecer la activación del NF-κB, esto a través del complejo NF-κB/Notch1, lo que incrementa el daño.
Citoplasmáticamente, las ERO generadas a partir de diversas fuentes, como la actividad de la NADPH oxidasa, activan diversas proteínas relacionada con el daño hepático como ERK1/2, p38 y JAK; estas promueven la activación de factores de transcripción como el AP1 y STAT3, modulando positivamente la expresión génica de citocinas pro y antiinflamatorias, respectivamente.
La señalización celular relacionada con la respuesta hepatoprotectora también se ve alterada por las ERO; las vías inducidas por la AMPK y el Nrf2/ARE se ven inhibidas por la acción de las especies oxidantes.
Las flechas verdes indican promoción de la respuesta, mientras que las rojas indican inhibición de la respuesta.
AMPK: proteínas cinasas activadas por adenosil monofosfato; ARE: elementos de respuesta antioxidante; ERO: especies reactivas de oxígeno; ERK1/2: cinasas reguladas por la señalización extracelular 1/2; JAK: cinasas Janus; LPS: lipopolisacárido; NF-κB: factor nuclear κB; Nrf2: factor relacionado al factor nuclear eritroidal 2.
Si bien son múltiples los factores involucrados en la generación de estrés oxidativo durante la enfermedad hepática alcohólica, el género y la idiosincrasia son puntos que deben ser tomados en cuenta.
Las mujeres desarrollan la enfermedad hepática por alcohol de forma más severa, con un consumo menor y en menos años de exposición a este hepatotóxico; los mecanismos exactos se desconocen pero algunas evidencias indican que las hormonas sexuales desempeñan un papel importante84. Mujeres con alcoholismo crónico tienen desórdenes en sus ciclos menstruales, incluyendo amenorrea, anovulación o ciclos irregulares y disfunción en la fase lútea85,86. Estudios hechos en ratas han demostrado que la ingestión de alcohol permite niveles bajos de estrógenos y ciclos anovulatorios caracterizados por una disminución de progesterona87; respuestas similares se han observado en mujeres alcohólicas premenopáusicas. Estos resultados indican que los estrógenos (y progesterona) son importantes en el desarrollo de la enfermedad hepática por alcohol y el impacto de las hormonas puede diferenciar dependiendo del estado en la vida de la mujer (menopausia, post menopausia), teniendo la ingesta de alcohol un papel importante en la generación del daño.
Por otro lado, todas las fases que participan en el desarrollo de la enfermedad hepática alcohólica están reguladas por genes, que ya sea solos o en combinación representan riesgos e influyen las reacciones con el etanol88. Después de su absorción, el etanol es degradado en el hígado y otros tejidos por la enzima alcohol deshidrogenasa (ADH) en el citosol y por el CYP2E1 en los microsomas. Diferentes clases de ADH son conocidas y entre los genes de la ADH humana conocidos, 2 son polimórficos con 3 alelos existentes para cada uno de ellos, el ADH2 y el ADH3, los cuales revelan características enzimáticas importantes; los alelos para ADH2 son ADH2*1 encontrado en los caucásicos y ADH2*2 en los asiáticos. De acuerdo con las diferencias en la capacidad para metabolizar alcohol en acetaldehído, se ha especulado que en los individuos con el alelo ADH2*2 hay un mayor riesgo para desarrollar daño hepático debido a una mayor exposición a acetaldehído89. Una situación similar se tiene con las variantes genéticas del CYP2E1, el alelo mutante CYP2E1 c2 está asociado con una elevada transcripción génica, los niveles de proteína y actividad enzimática comparadas con las del alelo c1 y esto puede resultar en una mayor exposición del hígado hacia el acetaldehído y a las ERO, exacerbando el daño hepático88.
Defensas antioxidantes en la enfermedad hepática alcohólicaDurante el consumo de etanol, se activan diversas vías que conllevan a la sobreproducción de ERO; sin embargo, esta producción masiva muchas veces sobrepasa la capacidad que tienen las defensas antioxidantes endógenas para poder contrarrestar el daño generado; a este fenómeno se le conoce como estrés oxidativo.
Múltiples son los mecanismos con los que el organismo cuenta para hacer frente a este fenómeno; el sistema de defensa antioxidante está constituido por un grupo de sustancias que retrasan o previenen la oxidación de un sustrato oxidable como los hidratos de carbono, proteínas, lípidos y moléculas del ADN90.
Se sabe que la administración de agentes antioxidantes, o agentes quelantes de hierro, cinc y cobre, así como agentes que reponen el glutatión reducido (GSH), como la N-acetil cisteína, puede aminorar o prevenir los efectos tóxicos del alcohol91. El reemplazo de lípidos poliinsaturados (necesarios para que se lleve a cabo la peroxidación lipídica) por triglicéridos de cadena mediana o lípidos saturados en la dieta administrados intragástricamente a ratas previene o disminuye la peroxidación de lípidos y el daño hepático inducido por el consumo de alcohol92,93. De manera interesante, la administración de antioxidantes como ebseleno, vitamina E, superóxido dismutasa y precursores de GSH previenen el daño hepático inducido por alcohol en ratas94.
Otros compuestos que han mostrado tener respuestas benéficas ante el daño hepático inducido por consumo de etanol son los corticoides; este grupo de fármacos pueden aminorar la respuesta inflamatoria durante la hepatitis alcohólica, disminuye la producción de citocinas, suprime la formación de aductos de acetaldehído e inhibe la producción de colágeno95. Existen evidencias de que los pacientes con una severa enfermedad hepática alcohólica y que son tratados con esteroides muestran mejoras histológicas en un corto plazo; esta mejoría está asociada a una disminución en circulación del TNF-α y en la expresión de la molécula de adhesión intercelular-1, así como cambios en la molécula de adhesión intercelular soluble-1 en la vena hepática96.
Sin duda, la planta más estudiada en el tratamiento de las enfermedades hepáticas es la Silybum marianum; esta consiste de 4 isómeros, que son la silibinina, isosilibina, silidianina y la silicristina. De todos ellos, la silibinina es el compuesto más activo. La silimarina ofrece una adecuada protección de acuerdo con los resultados de diferentes estudios de daño hepático experimental, ejerciendo buenos efectos antioxidantes97 y efectos antifibróticos98, además de ser un excelente inmunomodulador y estabilizador de las membranas celulares, así como en el proceso de regeneración98-101. Clínicamente, la silimarina ha demostrado tener efectos positivos en la enfermedad hepática alcohólica, la cirrosis hepática, el daño hepático inducido por el consumo de fármacos y además en pacientes diabéticos101.
La metadoxina (piridoxol L-2-pirrolidona-5-carboxilato) es otro de los compuestos que ha demostrado una gran eficacia en el tratamiento de la intoxicación aguda por alcohol; esta es el par iónico entre la pirrolidona carboxilato y la piridoxina. El carboxilato de pirrolidona participa en el metabolismo de los aminoácidos a través de la vía del glutatión102, participa en la síntesis de novo de ATP103 y previene la disminución de los niveles de ATP, tanto en el cerebro como en el hígado de ratas intoxicadas de manera aguda con etanol104. De manera importante, se sabe que la piridoxina incrementa el índice de degradación metabólica del etanol; además, reduce el daño en la función celular causada por el acetaldehído105. La metadoxina parece ser capaz de acelerar el metabolismo del etanol tanto en ratas como en humanos a través de favorecer el incremento de la actividad de la enzima acetaldehído deshidrogenasa, de aumentar el aclaramiento plasmático de etanol y acetaldehído, y la eliminación renal de las cetonas producto de su metabolismo106-108. Este compuesto ha sido capaz de prevenir la depleción de GSH, el daño oxidativo a nivel de lípidos, la acumulación de colágeno y la secreción de TNF-α inducida por el alcohol y el acetaldehído, tanto en hepatocitos como en células estelares109.
Además, la metadoxina ha demostrado efectos benéficos sobre la mortalidad temprana en pacientes con hepatitis alcohólica en el Hospital General de México (Higuera de la Tijera, datos no publicados).
Los compuestos antes mencionados son, en nuestra consideración, los que han destacado en el tratamiento de la enfermedad hepática alcohólica y muchos de ellos tienen suficiente evidencia científica en modelos animales de su eficacia; esto alienta a seguir estudiando a estas moléculas pero ahora en estudios clínicos controlados y así poder considerarlos en el futuro como una herramienta para el tratamiento farmacológico de esta enfermedad.
ConclusionesEl consumo crónico de etanol desempeña un papel relevante en la generación de la enfermedad hepática. Tomando en cuenta que, de manera natural durante los procesos bioquímicos, existe una biosíntesis normal de ERO, se considera que la ingestión de etanol favorece el estrés oxidativo y la promoción del daño inflamatorio y fibrótico. Debido a que son múltiples los mecanismos a través de los cuales el alcohol favorece la generación del daño hepático, la complejidad para desarrollar estrategias terapéuticas para el tratamiento de esta enfermedad es mayor y esto explica por qué actualmente aún no existe un tratamiento efectivo para poder resolver este problema de salud. Sin embargo, pensamos que al bloquear una de las vías de daño a través del empleo de antioxidantes, suplementos que favorezcan la generación de novo de GSH y agentes polifenólicos como la vitamina E o la silimarina, así como el empleo de fármacos como la metadoxina, podrían mejorar la calidad de vida de estos pacientes.
FinanciaciónLa Dra. Marina Galicia Moreno contó con el apoyo del Programa de Becas posdoctorales de la UNAM, DGAPA 2013-2014, así como del programa PAPIIT IA203113.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés.

