
La sospecha de síndrome de Lynch sin mutación conocida (SSL) se diagnostica cuando existe déficit de expresión de las proteínas reparadoras de ADN pero con estudio genético normal. El comportamiento y el manejo son controvertidos. Presentamos las características de pacientes con SSL y proponemos una vigilancia.
Material y métodosSe realiza análisis inmunohistoquímico (IMH) en familias con sospecha de síndrome de Lynch. Si existe pérdida de expresión, sin mutación BRAF, se procede al análisis germinal.
ResultadosDe ciento cuarenta y ocho pacientes en los que se realizó IMH, 23 presentaron pérdida de expresión. Siete fueron identificados como SSL: 3con cáncer de colon, 2con tumor endometrial y otros 2sanos con familiar afectado. La edad media fue de 56.9 años y solo uno presentó otro tumor asociado al síndrome de Lynch.
ConclusionesHasta que conozcamos mejor la etiología de esta entidad heterogénea, una vigilancia intermedia sería una estrategia adecuada.
Lynch-like syndrome is diagnosed when there is an expression deficit in DNA mismatch repair proteins but a normal genetic study. The behavior and management of that pathology are currently a subject of debate. We present herein the characteristics of patients with Lynch-like syndrome, together with a surveillance proposal.
Materials and methodsImmunohistochemistry was carried out on families suspected of presenting with Lynch syndrome. Germline analysis was done if there was loss of mismatch repair protein expression and no BRAF mutation.
ResultsOf the 148 patients that underwent immunohistochemistry testing, 23 presented with loss of mismatch repair protein expression. Seven of those patients were identified as having Lynch-like syndrome: 3had colon cancer, 2had endometrial tumor, and 2were healthy, with an affected relative. Mean patient age was 56.9 years and only one patient presented with another tumor associated with Lynch syndrome.
ConclusionsUntil there is a better understanding of the etiology of that heterogeneous entity, intermediate surveillance is an adequate strategy.
El síndrome de Lynch (SL) es la causa más frecuente de cáncer colorrectal (CCR) hereditario. Se debe a mutaciones germinales de los genes que corrigen los errores de emparejamiento durante la replicación del ADN (MLH1, MSH2, MSH6 y PMS2) o por la deleción del gen EPCAM que resulta en el silenciamiento de MSH2. La estrategia clásica para el diagnóstico de SL consiste en realizar el estudio genético a pacientes con criterios clínicos que presenten inestabilidad de microsatélites (IMS) alta o pérdida de expresión proteica en el estudio inmunohistoquímico (IMH). Sin embargo, esta práctica dejaría muchos pacientes con SL sin diagnosticar, por lo que se ha propuesto ampliar el uso de estas técnicas a todos los pacientes con CCR1.
Independientemente de la estrategia a utilizar, la generalización del uso de técnicas de inmunohistoquímica o moleculares hace que cada vez con mayor frecuencia nos encontremos resultados discordantes entre dichas pruebas y el estudio genético. Surge así el término CCR hereditario no polipósico (CCHNP), anteriormente intercambiable con el de SL, y que actualmente engloba un amplio espectro de entidades que presentan características clínicas similares al SL pero sin necesidad de cursar con las mutaciones germinales de los genes implicados en el SL. Los síndromes incluidos en el CCHNP pueden ser distribuidos en función de si presentan o no alteración en el sistema reparador de ADN demostrado bien por presencia de IMS alta o por la pérdida de expresión proteica con técnicas de inmunohistoquímica. Las condiciones que clínicamente cursan como SL pero en las que no se demuestran alteraciones en cualquiera de estas técnicas pueden ser atribuibles a la entidad conocida como CCR tipo X. También se incluiría dentro de este grupo las poliposis producidas por mutaciones en los dominios exonucleasa de los genes POLE y POLD12. Por otra parte, los pacientes con clínica compatible de SL, y en los que se objetiva alteración del sistema reparador de ADN, se incluyen 2entidades: el SL y la sospecha de SL sin mutación conocida (SSL) o «Lynch-like syndrome». Estos últimos se caracterizan por la ausencia de mutaciones germinales en los genes reparadores a pesar de presentar IMS alta o pérdida de expresión proteica en el tumor. La etiopatogenia, así como el seguimiento de estos pacientes, está aún por definir. La distinción de los diferentes síndromes que incluye el CCHNP es clínicamente relevante, porque la vigilancia de los pacientes y familiares a riesgo difiere según el riesgo de neoplasias colónicas o extracolónicas asociadas a cada entidad.
Presentamos el proceso diagnóstico, el comportamiento clínico y fenotípico de varios pacientes con SSL y realizamos una revisión de la literatura para proponer una estrategia de vigilancia adecuada.
Material y métodosSe analizan los pacientes con SSL diagnosticados entre enero del 2016 y junio del 2017 en la consulta de alto riesgo de un hospital de la red sanitaria pública de la comunidad de Madrid. A esta unidad se derivan pacientes y familiares con mayor riesgo de presentar CCR con base en su historia personal y familiar. En la consulta se procede a la identificación, el seguimiento y el consejo genético de pacientes con síndromes hereditarios digestivos. Para identificar a pacientes con SL se valoran los datos referentes a la estructura y antecedentes de la familia relacionados con enfermedades neoplásicas asociadas a dicho síndrome de al menos 3generaciones. En los pacientes donde se cumplen los criterios de Ámsterdam II3 o de Bethesda4 definidos, se investiga la existencia de alteración en el sistema reparador de ADN. Para ello, se evalúa sobre el tumor del paciente o familiar afectado, la expresión de las proteínas implicadas con técnicas de inmunohistoquímica, utilizando el sistema de visualización EnvisionTM (Dako, Región Capital, Dinamarca) y los anticuerpos prediluidos: MLH1 (ES05, Dako); MSH2 (FE11, Dako), MSH6 (EP49, Dako) y PMS2 (EP51, Dako). La interpretación se llevó a cabo bajo microscopio Nikon eclipse e400 (Nikon, Ámsterdam, Holanda), a 10 y 20 aumentos. La deficiencia de estas proteínas se define como pérdida completa de expresión nuclear en las células tumorales con controles internos positivos. En las muestras en las que se objetivó pérdida de expresión de MLH1, se analiza la mutación V600E (1799 T>A) del gen BRAF con técnicas moleculares (Cobas® 4800 [Roche Diagnostics, Mannheim, Alemania).
En caso de ausencia de expresión de alguna proteína, no justificada por metilación de MLH1, se realiza el análisis germinal de los 5genes involucrados en el SL mediante enriquecimiento y secuenciación en plataformas de alto rendimiento. Si no se encuentran mutaciones patogénicas, se incluyen como pacientes con SSL.
Las variables demográficas y clínicas de estos pacientes se recogen mediante historia clínica dirigida. Las variables endoscópicas se obtienen de los informes de las colonoscopias desde el programa departamental Endobase® (Olympus, Hamburgo, Alemania). Todas las colonoscopias se realizan por facultativos especialistas de digestivo con endoscopios de luz blanca EC-380LKp (Pentax, Tokio, Japón) y CF-Q145L (Olympus, Tokio, Japón).
Análisis estadísticoSe realiza un análisis descriptivo de todas las variables recogidas en el estudio. Las variables cualitativas se describieron como medida de tendencia central acompañadas de su medida de dispersión según fuera la distribución de la variable. Las variables cualitativas se describieron mediante frecuencias relativas.
ResultadosDe los setecientos noventa y siete pacientes remitidos a la consulta durante el periodo de estudio, 434 (54.5%) fueron dirigidos por antecedentes familiares o con diagnóstico de CCR con sospecha de síndrome hereditario. De ellos, 211 (48.6%) presentaban criterios clínicos de SL (20 de Ámsterdam II y 197 de Bethesda). En 63 pacientes no se pudo realizar estudio IMH por haber fallecido el familiar afectado o haber pasado mucho tiempo desde la intervención quirúrgica para recuperar la pieza tumoral.
En un total de 148 pacientes (60.1% mujeres), con una edad media de 54.6±13.3 años, se realizó estudio IMH sobre el tumor del paciente (n=71) o sobre el de algún familiar afectado (n=77).
En 29 se obtuvo pérdida de expresión proteica con evidencia de mutación BRAF en 6 de los que presentaron ausencia de MLH1. De los 23 pacientes restantes, 21 se realizaron estudio genético mientras que 2los rechazaron. El análisis fue normal en 12 de ellos, en 5no se puede descartar por completo un SL por realizarse el estudio IMH sobre tumor del familiar y el estudio genético en el familiar a riesgo. En estos casos, un resultado verdaderamente negativo solo se podría establecer si otro familiar en riesgo presentara un resultado positivo.
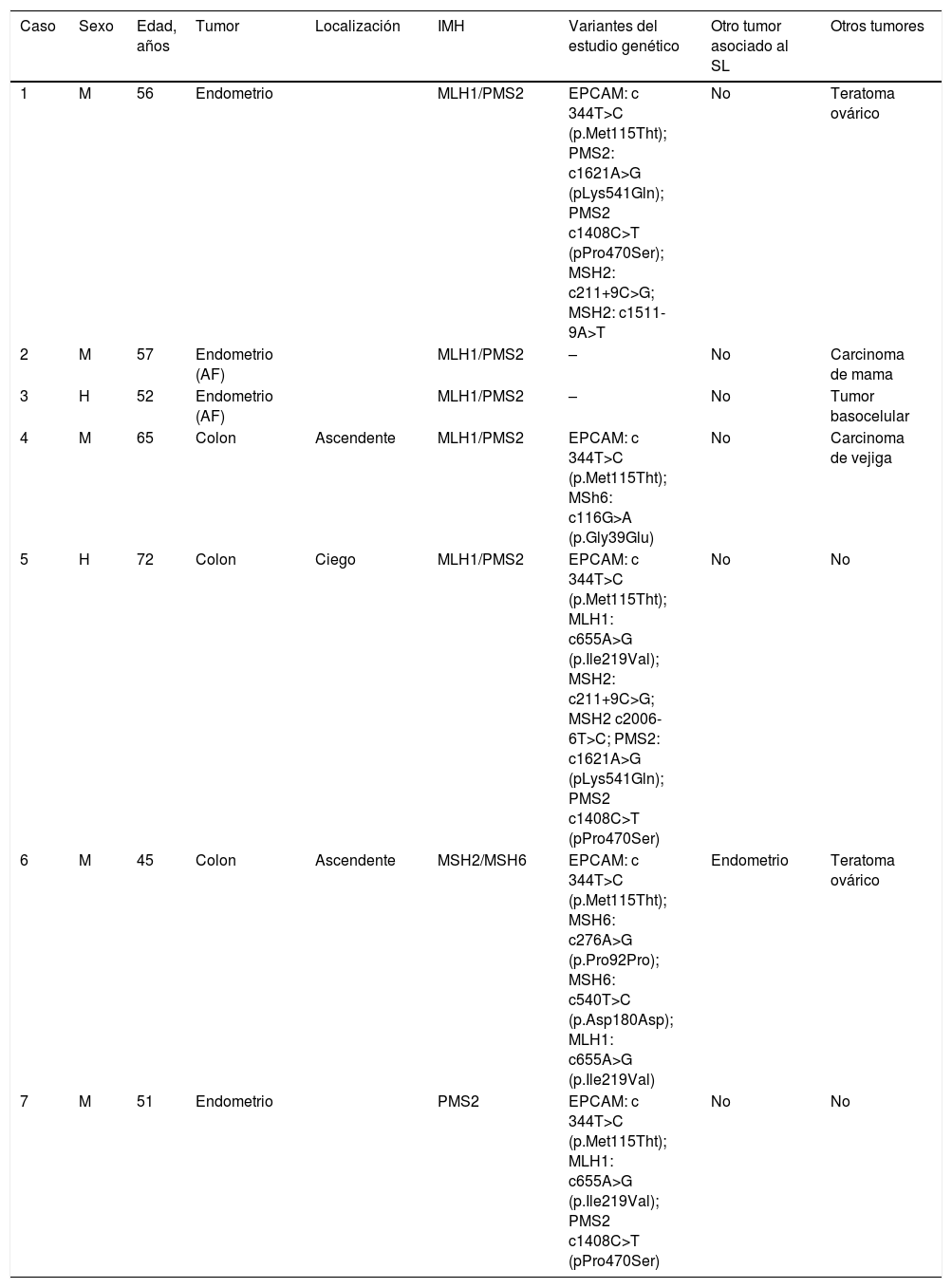
Finalmente, un total de 7 pacientes (33.3%) cumplen criterios de SSL. Las características clínicas, moleculares y genéticas de estos pacientes se resumen en la tabla 1. Cinco son pacientes afectados, mientras que los otros 2son sujetos sanos familiares de pacientes con SSL. La edad media fue de 56.9±9 años, con una proporción mayor de mujeres (5/2).
Características clínicas, moleculares y genéticas de los pacientes con sospecha de síndrome de Lynch sin mutación conocida
| Caso | Sexo | Edad, años | Tumor | Localización | IMH | Variantes del estudio genético | Otro tumor asociado al SL | Otros tumores |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 56 | Endometrio | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); PMS2: c1621A>G (pLys541Gln); PMS2 c1408C>T (pPro470Ser); MSH2: c211+9C>G; MSH2: c1511-9A>T | No | Teratoma ovárico | |
| 2 | M | 57 | Endometrio (AF) | MLH1/PMS2 | – | No | Carcinoma de mama | |
| 3 | H | 52 | Endometrio (AF) | MLH1/PMS2 | – | No | Tumor basocelular | |
| 4 | M | 65 | Colon | Ascendente | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); MSh6: c116G>A (p.Gly39Glu) | No | Carcinoma de vejiga |
| 5 | H | 72 | Colon | Ciego | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); MLH1: c655A>G (p.Ile219Val); MSH2: c211+9C>G; MSH2 c2006-6T>C; PMS2: c1621A>G (pLys541Gln); PMS2 c1408C>T (pPro470Ser) | No | No |
| 6 | M | 45 | Colon | Ascendente | MSH2/MSH6 | EPCAM: c 344T>C (p.Met115Tht); MSH6: c276A>G (p.Pro92Pro); MSH6: c540T>C (p.Asp180Asp); MLH1: c655A>G (p.Ile219Val) | Endometrio | Teratoma ovárico |
| 7 | M | 51 | Endometrio | PMS2 | EPCAM: c 344T>C (p.Met115Tht); MLH1: c655A>G (p.Ile219Val); PMS2 c1408C>T (pPro470Ser) | No | No |
AF: antecedentes familiares; H: hombre; IMH: proteína no expresada en inmunohistoquímica; M: mujer; SL: síndrome de Lynch.
De los pacientes afectados con SSL, 3presentaron CCR y otros 2tumores endometriales. En 3se objetiva pérdida de expresión de MLH1/PMS2 con BRAF no mutado, en uno de MSH2/MSH6 y en otro ausencia aislada de PMS2. Destaca así mismo que todos los CCR fueron de localización proximal.
En todos los pacientes se han realizado las pruebas de vigilancia del SL sin encontrar otros tumores asociados, salvo una paciente que presentó también adenocarcinoma endometrial.
DiscusiónLa prevalencia de SSL varía del 56 al 71% en las cohortes de CCR5-7 y entre el 30-64% en los tumores endometriales8,9. En nuestra serie, el porcentaje fue algo menor (33.3%).
La causa es desconocida si bien se han descrito varias teorías: mutaciones germinales de estos genes que no detectemos con las técnicas actuales2; mutaciones somáticas de MLH1 y MSH2 que produzcan inactivación de estos genes10,11; y mutaciones en otros genes diferentes de los genes reparadores de ADN2.
Poco se sabe sobre el riesgo de CCR en estos pacientes. En un estudio se obtiene que la incidencia de CCR en los familiares de pacientes con SSL fue menor que en los pacientes con SL (tasa de incidencia estandarizada [TIE] de 6.04, IC del 95%, 3.58-9.54 para el SL, y del 2.12 en SLL; IC del 95%, 1.16-3.56, p <0.01) pero mayor que en los pacientes con CCR esporádico (TIE 0.48, IC del 95% 0.27-0.79, p <0.001)6. En otro estudio se obtienen resultados similares, con un riesgo inferior en los pacientes con SSL que los pacientes con SL (RR en SL: 5.37; IC del 95%, 4.16-6.94, y RR en SSL: 2.06; IC del 95%, 1.59-2.67, p <0.001) pero superior a las familias de pacientes con CCR esporádico (RR 1.04, IC del 95%, 0.82-1.31)7.
Con respecto a tumores extracolónicos asociados al SL, en el estudio antes referido no se encuentra un mayor riesgo en pacientes con SSL, si bien puede atribuirse al bajo de número de casos detectados6. En un reciente trabajo, los antecedentes familiares de tumores asociados al SL en pacientes con tumor gástrico con SSL fueron mayores que en el grupo de cáncer esporádico (76.5% vs. 38.6%, p=0.004)12.
Los pacientes con SSL presentan riesgo de CCR a edades precoces, similar a los pacientes con SL (58-54 vs. 49 años)6,7. También la proporción de pacientes con tumor endometrial antes de los 50 años es mayor en el SSL que en el tumor esporádico (23.5% vs. 14.1%)12.
No existe suficiente evidencia para definir un seguimiento de estos pacientes. Dado que el riesgo de CCR es intermedio entre las familias con SL y con CCR esporádico, una estrategia adecuada sería realizar un seguimiento intermedio con intervalos de 2-3 años, en función de la historia familiar, comenzando a una edad parecida a la de los pacientes con SL.
No obstante, los pacientes con SSL constituyen un grupo heterogéneo en el que posiblemente estén incluidos desde pacientes con verdadero SL a pacientes con CCR esporádico. Cuando conozcamos más los mecanismos etiopatogénicos, es probable que estos pacientes se reclasifiquen en entidades diferenciadas y podamos ofrecerles una vigilancia individualizada.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónLos autores no recibieron financiamiento para este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de interés para la realización de este trabajo.

