
Lynch-like syndrome is diagnosed when there is an expression deficit in DNA mismatch repair proteins but a normal genetic study. The behavior and management of that pathology are currently a subject of debate. We present herein the characteristics of patients with Lynch-like syndrome, together with a surveillance proposal.
Materials and methodsImmunohistochemistry was carried out on families suspected of presenting with Lynch syndrome. Germline analysis was done if there was loss of mismatch repair protein expression and no BRAF mutation.
ResultsOf the 148 patients that underwent immunohistochemistry testing, 23 presented with loss of mismatch repair protein expression. Seven of those patients were identified as having Lynch-like syndrome: 3had colon cancer, 2had endometrial tumor, and 2were healthy, with an affected relative. Mean patient age was 56.9 years and only one patient presented with another tumor associated with Lynch syndrome.
ConclusionsUntil there is a better understanding of the etiology of that heterogeneous entity, intermediate surveillance is an adequate strategy.
La sospecha de síndrome de Lynch sin mutación conocida (SSL) se diagnostica cuando existe déficit de expresión de las proteínas reparadoras de ADN pero con estudio genético normal. El comportamiento y el manejo son controvertidos. Presentamos las características de pacientes con SSL y proponemos una vigilancia.
Material y métodosSe realiza análisis inmunohistoquímico (IMH) en familias con sospecha de síndrome de Lynch. Si existe pérdida de expresión, sin mutación BRAF, se procede al análisis germinal.
ResultadosDe ciento cuarenta y ocho pacientes en los que se realizó IMH, 23 presentaron pérdida de expresión. Siete fueron identificados como SSL: 3con cáncer de colon, 2con tumor endometrial y otros 2sanos con familiar afectado. La edad media fue de 56.9 años y solo uno presentó otro tumor asociado al síndrome de Lynch.
ConclusionesHasta que conozcamos mejor la etiología de esta entidad heterogénea, una vigilancia intermedia sería una estrategia adecuada.
Lynch syndrome (LS) is the most frequent cause of inherited colorectal cancer (CRC). It is due to germline mutations of the genes that correct mismatch errors during DNA replication (MLH1, MSH2, MSH6, and PMS2), or to the EPCAM gene deletion that results in MSH2 silencing. The classic strategy for LS diagnosis is the genetic testing of patients with clinical criteria who present with high microsatellite instability (MSI) or loss of protein expression in immunohistochemistry (IHC) testing. However, that practice has left many patients with LS undiagnosed, thus the broadened use of those techniques has been proposed to include all patients with CRC.1
Regardless of the strategy utilized, the generalized use of immunohistochemical or molecular techniques has increasingly produced discordant results between those tests and genetic screening. Thus, the term hereditary nonpolyposis colorectal cancer (HNPCC) arose. It was previously interchangeable with LS, but today it encompasses a broad spectrum of entities that present characteristics similar to LS, but do not have the germline mutations of the genes involved in LS. The syndromes included in HNPCC can be distributed in relation to whether or not they present an alteration in the DNA repair system that is demonstrated by the presence of high MSI or the loss of mismatch repair protein expression through immunohistochemical techniques. The conditions that clinically present as LS, but with no alterations identified by any of those techniques, can be attributable to the entity known as CRC type X. Polyposis produced by mutations in the exonuclease domains of the POLE and POLD1 genes are also included in that group2. Furthermore, 2 entities are included in patients with clinical symptoms consistent with LS and DNA repair system alterations: LS and suspected LS with no known mutation, or the so-called “Lynch-like syndrome” (LLS). LLS is characterized by the absence of germline mutations in the repair genes, despite the presence of high MSI or protein expression loss in the tumor. Both the disease etiopathogenesis and follow-up of those patients have yet to be defined. Distinguishing between the different syndromes included in HNPCC is clinically relevant, because surveillance of the patients and their at-risk relatives differs according to the risk for colonic or extracolonic neoplasias associated with each entity.
We present herein the diagnostic process and clinical and phenotypic behavior of various patients with LLS, as well as a literature review, to propose an adequate surveillance strategy.
Materials and methodsPatients diagnosed with LLS at the high-risk consultation of a community public healthcare hospital in Madrid within the time frame of January 2016 and June 2017 were analyzed. Patients and their relatives at high risk for CRC, based on their personal and family histories, are seen at that unit. At consultation, patients with hereditary gastrointestinal syndromes are identified, followed, and given genetic counseling. To identify patients with LS, data related to family structure and a family history of neoplastic diseases associated with LS going back at least 3 generations are evaluated. Alterations in the DNA repair system are searched for in patients that meet the Amsterdam II3 or Bethesda4 criteria. The tumor of the patient or affected relative is evaluated and immunohistochemistry is performed to determine expression of the proteins involved, utilizing the EnvisionTM (Dako, Capital Region, Denmark) visualization system and the prediluted antibodies: MLH1 (ES05, Dako); MSH2 (FE11, Dako), MSH6 (EP49, Dako), and PMS2 (EP51, Dako). Interpretation was carried out using a Nikon Eclipse e400 microscope (Nikon, Amsterdam, Netherlands), at magnifications of x10 and x20. The deficiency of those proteins was defined as complete loss of nuclear expression in the tumor cells with positive internal controls. In the samples with MLH1 expression loss, the V600E (1799 T>A) mutation of the BRAF gene was analyzed through molecular techniques (Cobas® 4800 [Roche Diagnostics, Mannheim, Germany]).
When the absence of expression of a protein was not justified by MLH1 methylation, the germlines of the 5 genes involved in LS were analyzed through high performance enrichment and sequencing platforms. If no pathogenic mutations were found, the samples were included as patients with LLS.
The demographic and clinical variables of those patients were collected through targeted clinical histories. The endoscopic variables were obtained from the colonoscopy reports, utilizing the departmental Endobase® (Olympus, Hamburg, Germany) program. All the colonoscopies were performed by gastrointestinal specialists, with EC-380LKp (Pentax, Tokyo, Japan) and CF-Q145L (Olympus, Tokyo, Japan) white light endoscopes.
Statistical analysisA descriptive analysis of all the variables collected in the study was carried out. The qualitative variables were described as measures of central tendency, accompanied by their dispersion measure, according to the distribution of the variable. The qualitative variables were described through relative frequencies.
ResultsOf the seven hundred and ninety-seven patients seen in consultation within the study time frame, 434 (54.5%) were referred because of family histories or for CRC diagnosis with suspicion of a hereditary syndrome. Of those patients, 211 (48.6%) presented with clinical criteria of LS (20 with the Amsterdam II criteria and 197 with the Bethesda criteria). IHC study could not be done on 63 patients, either because the affected relative had died or too much time had passed since the surgical intervention to be able to recover the tumor specimen.
From the total of 148 patients (60.1% women) with a mean age of 54.6 ± 1.3 years, IHC was carried out on the tumor of the patient (n=71) or on the tumor of an affected relative (n=77).
There was protein expression loss in 29 patients, with evidence of BRAF mutation in 6 of the patients with absence of MLH1. Of the 23 remaining patients, 21 underwent genetic testing, whereas 2 rejected it. Genetic screening was normal in 12 of those patients, but LS could not be completely ruled out in 5 of them, because the IHC study was carried out on the tumor of a relative and the genetic study on an at-risk relative. In those cases, a true negative result could only be established if another at-risk relative presented with a positive result.
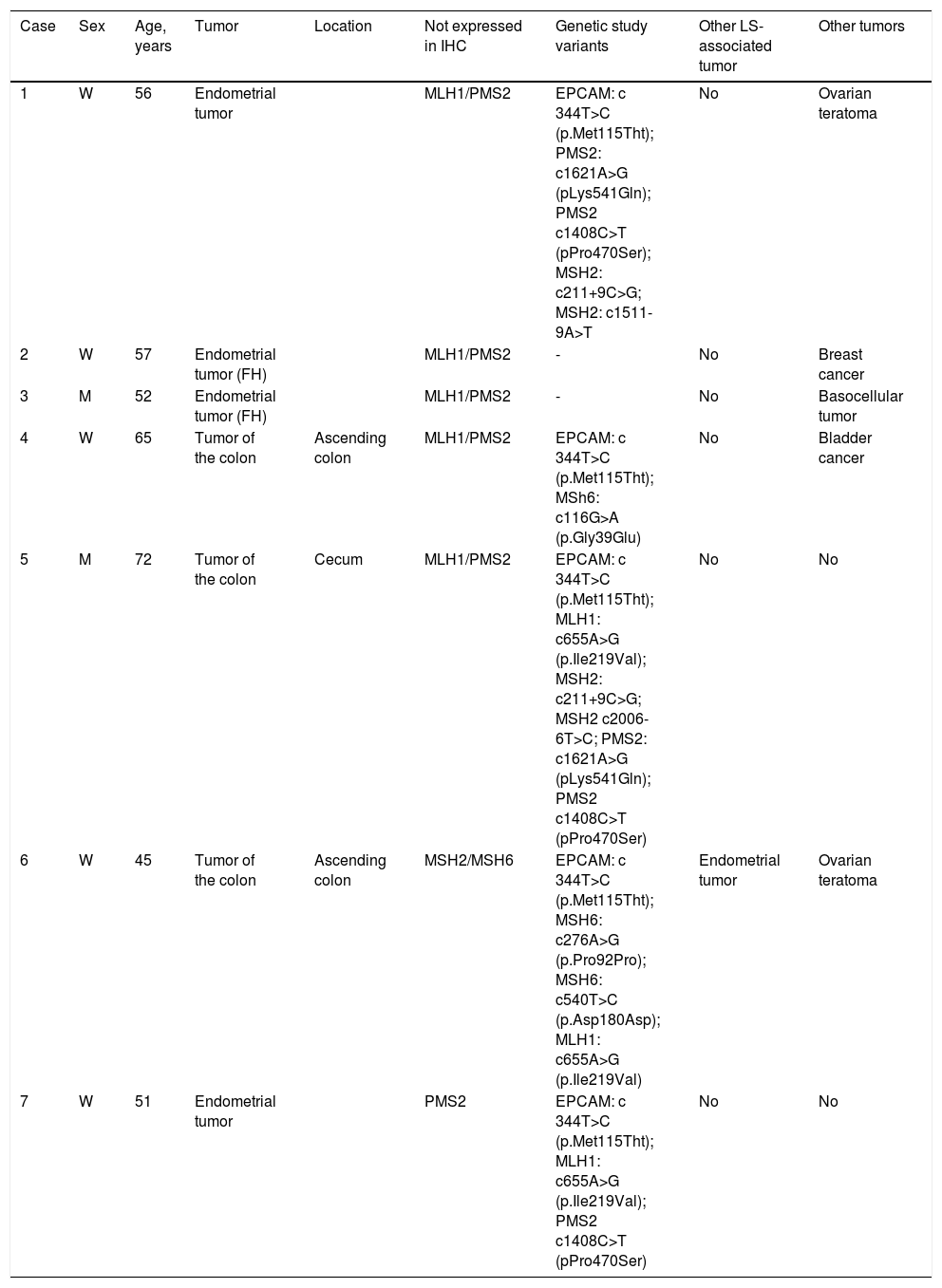
Finally, a total of 7 patients (33.3%) met the LLS criteria. The clinical, molecular, and genetic characteristics of those patients are shown in Table 1. Five of the patients were directly affected, whereas the other 2 were healthy subjects with relatives that had LLS. The mean age of the seven patients was 56.9 ± 9 years, with a higher number of women (5/2).
Clinical, molecular, and genetic characteristics of the patients with suspected Lynch syndrome with no known mutation.
| Case | Sex | Age, years | Tumor | Location | Not expressed in IHC | Genetic study variants | Other LS-associated tumor | Other tumors |
|---|---|---|---|---|---|---|---|---|
| 1 | W | 56 | Endometrial tumor | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); PMS2: c1621A>G (pLys541Gln); PMS2 c1408C>T (pPro470Ser); MSH2: c211+9C>G; MSH2: c1511-9A>T | No | Ovarian teratoma | |
| 2 | W | 57 | Endometrial tumor (FH) | MLH1/PMS2 | - | No | Breast cancer | |
| 3 | M | 52 | Endometrial tumor (FH) | MLH1/PMS2 | - | No | Basocellular tumor | |
| 4 | W | 65 | Tumor of the colon | Ascending colon | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); MSh6: c116G>A (p.Gly39Glu) | No | Bladder cancer |
| 5 | M | 72 | Tumor of the colon | Cecum | MLH1/PMS2 | EPCAM: c 344T>C (p.Met115Tht); MLH1: c655A>G (p.Ile219Val); MSH2: c211+9C>G; MSH2 c2006-6T>C; PMS2: c1621A>G (pLys541Gln); PMS2 c1408C>T (pPro470Ser) | No | No |
| 6 | W | 45 | Tumor of the colon | Ascending colon | MSH2/MSH6 | EPCAM: c 344T>C (p.Met115Tht); MSH6: c276A>G (p.Pro92Pro); MSH6: c540T>C (p.Asp180Asp); MLH1: c655A>G (p.Ile219Val) | Endometrial tumor | Ovarian teratoma |
| 7 | W | 51 | Endometrial tumor | PMS2 | EPCAM: c 344T>C (p.Met115Tht); MLH1: c655A>G (p.Ile219Val); PMS2 c1408C>T (pPro470Ser) | No | No |
FH: family history; IHC: protein not expressed in immunohistochemistry; LS Lynch syndrome; M: man; W: woman.
Of the patients with LLS, 3 presented with CRC and 2 had endometrial tumors. Three of the patients had loss of MLH1/PMS2 expression with unmutated BRAF, one patient had loss of MSH2/MSH6 expression, and one had an isolated absence of PMS2. It was striking that all of the CRCs had a proximal location.
LS surveillance tests were carried out in all the patients and no other tumors were found, except in one female patient that also presented with endometrial adenocarcinoma.
DiscussionThe prevalence of LLS varies from 56 to 71% in CRC cohorts5–7 and between 30-64% in endometrial tumors.8,9 In our case series, the percentage was somewhat lower (33.3%).
The cause of Lynch-like syndrome is unknown, albeit several theories have been described: germline mutations of the genes that are not detected by the current techniques;2 somatic MLH1 and MSH2 mutations that produce inactivation of those genes;10,11 and mutations in other genes different from the DNA repair genes.2
Little is known about the risk for CRC in those patients. Incidence of CRC in relatives of patients with LLS was reported in one study to be lower than that in patients with LS (standardized incidence ratio [SIR] of 6.04, 95% CI: 3.58-9.54 for LS and SIR of 2.12, 95% CI: 1.16-3.56 for LLS, p< 0.01) but higher than that in patients with sporadic CRC (SIR: 0.48, 95% CI: 0.27-0.79, p < 0.001).6 Results were similar in another study that reported a lower risk in patients with LLS than in patients with LS (RR in LS: 5.37; 95% CI: 4.16-6.94 and RR in LLS: 2.06; 95% CI: 1.59-2.67, p < 0.001) but higher than that of families of patients with sporadic CRC (RR: 1.04, 95% CI: 0.82-1.31).7
With respect to extracolonic tumors associated with LS, in the study mentioned above, no higher risk was found in patients with LLS, even though that could be attributed to the low number of cases detected.6 In a recent study, family histories of tumors associated with LS in patients with gastric cancer were higher in the LLS group than in the sporadic cancer group (76.5% vs. 38.6%, p = 0.004).12
Patients with LLS presented with risk for CRC at early ages, similar to the patients with LS (58-54 vs. 49 years).6,7 The number of patients with endometrial tumor before 50 years of age was also higher in the patients with LLS than in those with sporadic tumor (23.5% vs. 14.1%).12
There is not enough evidence to define follow-up in patients with Lynch-like syndrome. Given that there is intermediate risk for CRC in families with LS and families with sporadic CRC, an adequate strategy would be to carry out intermediate follow-up with 2 to 3-year intervals, in relation to family history, beginning at an age similar to that in patients with LS.
Nevertheless, patients with LLS make up a heterogeneous group that may include patients with true LS to patients with sporadic CRC. When more is understood about the etiopathogenic mechanisms, most likely those patients will be reclassified into differentiated entities and we can offer them individualized surveillance.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Adán-Merino L, Aldeguer-Martínez M, Alonso-Gamarra E, Valentín-Gómez F, Zaera-De la Fuente C, Martín-Chávarri S. Diagnóstico y comportamiento clínico de pacientes con sospecha de síndrome de Lynch sin mutación conocida. Revista de Gastroenterología de México. 2018;83:470–474.

