
La pancreatitis crónica es un proceso inflamatorio progresivo del páncreas que lleva a fibrosis, pérdida de la función exocrina y endocrina con cambios estructurales de evidencia radiológica o histológica1,2.
El curso clínico de la enfermedad se caracteriza por episodios recurrentes de pancreatitis aguda de aparición en edades tempranas, seguida de un estadio final con calcificaciones, insuficiencia pancreática y diabetes mellitus en la mayoría de los pacientes1.
La incidencia de la pancreatitis crónica por cualquier causa es de 3.5-10/100,000 habitantes/año en Europa y Estados Unidos3. Existe un significativo porcentaje de casos (10-25%) en los que no es posible definir su causa, obligando a la clasificación de pancreatitis crónica idiopática. En niños esta representa el 40-60% de los casos4.
En las últimas décadas, se han identificado alteraciones en otros genes además del regulador de conductancia transmembrana de fibrosis quistica (CFTR), como el tripsinógeno catiónico (PRSS1), tripsinógeno aniónico (PRSS2), inhibidor de serina proteasa tipo Kazal 1 (SPINK1) y quimiotripsina C
(CTRC), que desempeñan un papel importante en la fisiopatología de la pancreatitis crónica, disminuyendo el porcentaje de casos definidos previamente como idiopáticos4,5.
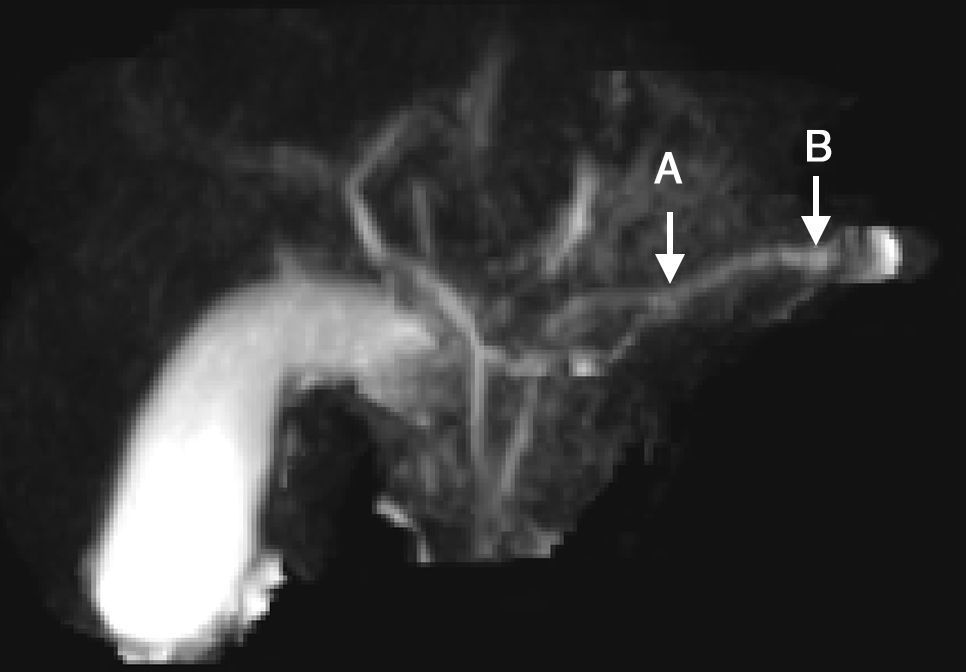
Presentamos el caso de un paciente que ingresa a los 8 años de edad al Hospital Pablo Tobón Uribe, por cuadro de dolor abdominal localizado en el epigastrio, náuseas y vómito. En adecuado estado nutricional, con dolor a la palpación del epigastrio, sin otros signos patológicos. Antecedente de un episodio previo de pancreatitis aguda a los 7 años y antecedente familiar de hermano con historia de pancreatitis aguda, sin otros datos relevantes. El cuadro de ingreso fue indicativo de pancreatitis aguda no complicada, con enzimas pancreáticas elevadas (amilasa 1,359; lipasa 3,098) (tabla 1). La resonancia magnética del páncreas realizada revela dilatación irregular del conducto de Wirsung (fig. 1). Teniendo en cuenta los antecedentes y los hallazgos bioquímicos y radiológicos, se inicia abordaje diagnóstico para pancreatitis crónica, descartándose otras causas, como hipercalcemia y pancreatitis autoinmune, con cuantificación de inmunoglobulina G subclase 4, que se encontró dentro de parámetros normales. Se realizaron estudios moleculares que incluyeron 60 mutaciones para el CFTR las cuales fueron negativas, reportando solo la presencia de la mutación N34S del gen SPINK 1, en el exón 3. En un año de seguimiento en la institución, el paciente presentó un nuevo cuadro de pancreatitis aguda no complicada y no ha presentado evidencia de insuficiencia endocrina o exocrina.
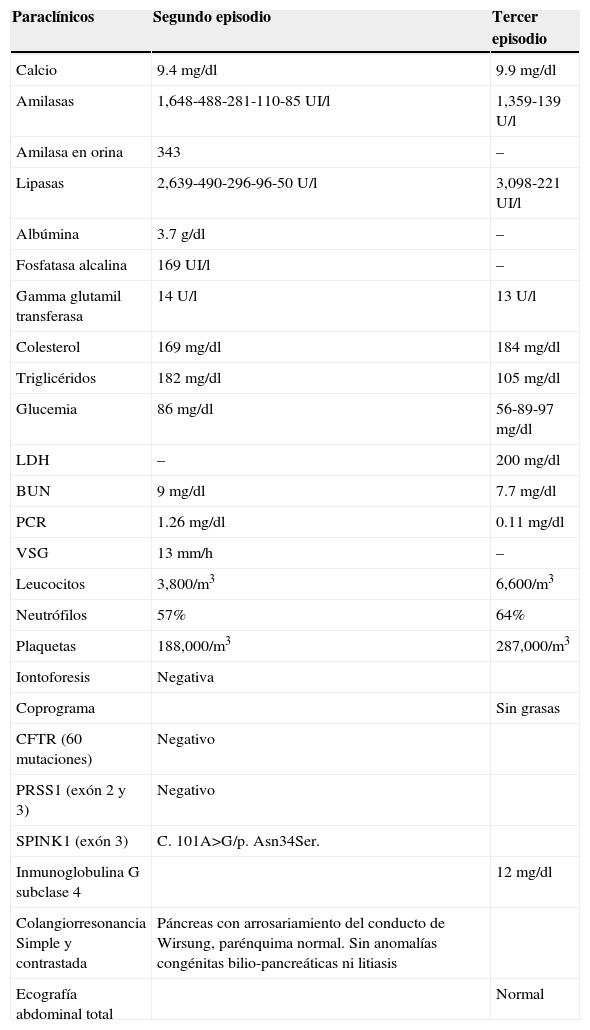
Estudios paraclínicos, de imagen y genéticos realizados
| Paraclínicos | Segundo episodio | Tercer episodio |
|---|---|---|
| Calcio | 9.4 mg/dl | 9.9 mg/dl |
| Amilasas | 1,648-488-281-110-85 UI/l | 1,359-139 U/l |
| Amilasa en orina | 343 | – |
| Lipasas | 2,639-490-296-96-50 U/l | 3,098-221 UI/l |
| Albúmina | 3.7 g/dl | – |
| Fosfatasa alcalina | 169 UI/l | – |
| Gamma glutamil transferasa | 14 U/l | 13 U/l |
| Colesterol | 169 mg/dl | 184 mg/dl |
| Triglicéridos | 182 mg/dl | 105 mg/dl |
| Glucemia | 86 mg/dl | 56-89-97 mg/dl |
| LDH | – | 200 mg/dl |
| BUN | 9 mg/dl | 7.7 mg/dl |
| PCR | 1.26 mg/dl | 0.11 mg/dl |
| VSG | 13 mm/h | – |
| Leucocitos | 3,800/m3 | 6,600/m3 |
| Neutrófilos | 57% | 64% |
| Plaquetas | 188,000/m3 | 287,000/m3 |
| Iontoforesis | Negativa | |
| Coprograma | Sin grasas | |
| CFTR (60 mutaciones) | Negativo | |
| PRSS1 (exón 2 y 3) | Negativo | |
| SPINK1 (exón 3) | C. 101A>G/p. Asn34Ser. | |
| Inmunoglobulina G subclase 4 | 12 mg/dl | |
| Colangiorresonancia Simple y contrastada | Páncreas con arrosariamiento del conducto de Wirsung, parénquima normal. Sin anomalías congénitas bilio-pancreáticas ni litiasis | |
| Ecografía abdominal total | Normal |
BUN: nitrógeno ureico; CFTR: regulador de conductancia transmembrana de fibrosis quistica; LDH: lactato deshidrogenasa; PCR: proteína C reactiva; PRSS1: tripsinógeno catiónico; SPINK1: inhibidor de serina proteasa tipo Kazal 1.; VSG: velocidad de sedimentación globular.
 Zona de estenosis focal. B) Dilatación. Imagen en arrosariamiento del conducto de Wirsung.")
El paciente descrito ha presentado 3 cuadros de pancreatitis aguda con periodos intercríticos asintomáticos, dilatación irregular del conducto de Wirsung evidenciada por imagen e identificación de la mutación N34S del gen SPINK1. Este gen mutado se ha asociado a pancreatitis aguda recurrente, crónica hereditaria, alcohólica, calcificante tropical e idiopática3. También se ha asociado a mayor tasa de litiasis pancreática, seudoquistes y calcificaciones4. De igual forma, se ha visto en un pequeño porcentaje de los controles asintomáticos y en otras patologías pancreáticas, lo cual indica que estas mutaciones, más que causales, son factores modificadores de la enfermedad. En este caso no se encontró otra causa atribuible más que la alteración del SPINK1 con la limitación que no se estudiaron otros genes, como CTRC, y otros exones del PRSS1.
La presentación clínica más frecuente es el inicio a edades tempranas, alrededor de los 10 años, con cuadros intermitentes de náuseas y vómitos, donde el dolor abdominal puede no ser el síntoma principal, con progresión lenta hacia la insuficiencia pancreática y el compromiso de la función endocrina, que puede ocurrir en edades más avanzadas, con evidencia radiológica de cambios estructurales (dilatación irregular de los conductos pancreáticos, atrofia, calcificación glandular, alteraciones en la grasa peripancreática)2,3,5,6.
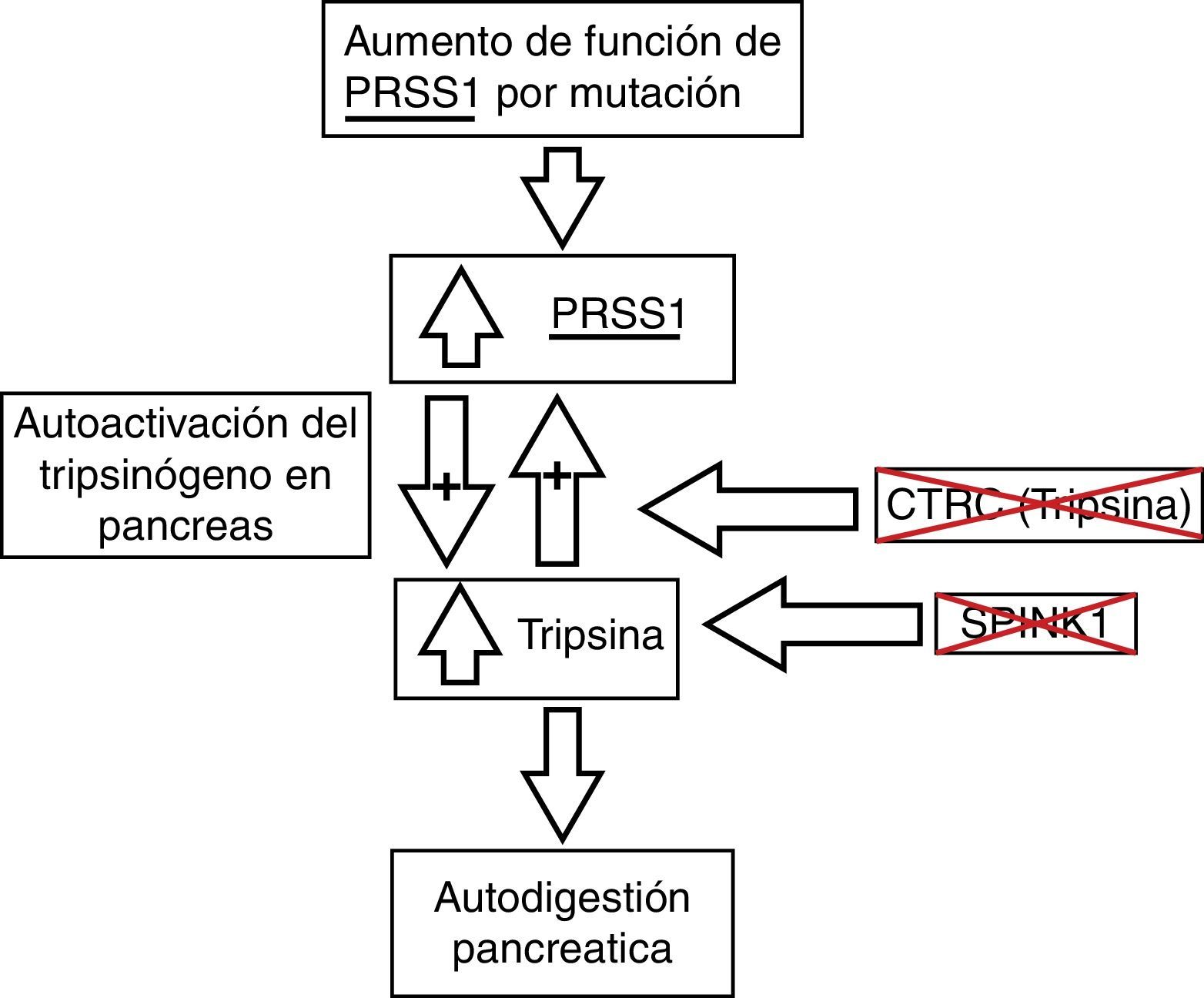
El proceso fisiopatológico de esta entidad aún es materia de investigación. Los hallazgos actuales señalan que es un proceso de autodigestión por la activación prematura del tripsinógeno secundario a mutaciones en los genes que codifican proteínas que se encargan de mantenerlo inactivo mientras se encuentra en el páncreas (fig. 2). Las alteraciones en el PRSS1 han sido descritas como las principales mutaciones asociadas a pancreatitis crónica; sin embargo, en los últimos años han emergido asociaciones con otros genes, como PRSS2, SPINK1 y CTRC7-9.

Fisiopatología de la pancreatitis crónica y sus bases genéticas. CTRC: quimiotripsina C; PRSS1: tripsinógeno catiónico; PRSS2: tripsinógeno aniónico; SPINK1: inhibidor de serina proteasa tipo Kazal 1. Adaptado de Derikx et al.7
La primera línea de bloqueo del tripsinógeno es el SPINK1, que es capaz de inhibir alrededor del 20% del total de la actividad de la tripsina dentro del páncreas. La mutación más comúnmente implicada de este gen es la N34S, según datos recientes de estudios genéticos de pacientes con pancreatitis crónica hereditaria e idiopática3,5,6,10.
La prevalencia de estas mutaciones varía de acuerdo con el área geográfica. En Latinoamérica se han publicado algunas series que reportan las mutaciones de estos genes en la población pediátrica con pancreatitis recurrente, siendo las más frecuentes la del gen PRSS1, no así la SPINK1-NS34, que ha sido encontrada en muy pocos casos11,12,13.
La pancreatitis crónica hereditaria es una entidad de baja prevalencia en la población pediátrica, con limitación en su diagnóstico por el bajo índice de sospecha y la dificultad para la identificación de mutaciones genéticas especificas en nuestro medio5, haciendo relevante la divulgación de casos clínicos como este, que evidencian las características típicas de esta patología y un abordaje diagnóstico completo, permitiendo la anticipación a posibles complicaciones, la prevención de factores de riesgo ambientales y el asesoramiento genético de la familia.
FinanciamientoNo se recibió financiación por la realización del escrito.
Conflicto de interesesLos autores no declaran conflicto de intereses.

