

Peutz-Jeghers syndrome is a rare autosomal dominant inherited disease caused by a germline mutation of the STK11/LKB1 gene, located on chromosome 19p13.3. It is characterized by mucocutaneous hyperpigmentation, hamartomatous polyposis, and predisposition to cancer. The aim of the present study was to identify and register patients with Peutz-Jeghers syndrome, describe the disease, and estimate its prevalence in Valencia (Spain).
Materials and methodsA print-out of the clinical histories from 10 hospitals was obtained utilizing the ICD-9 code 759.6 from the Minimum Basic Data Set of Hospital Admissions of the Spanish Ministry of Health and Consumer Affairs.
ResultsFrom a total of 405 clinical histories found, 15 (9 males and 6 females) fit the diagnostic criteria of Peutz-Jeghers syndrome. Mean age at diagnosis was 13.8 years and mean age at death was 54.2 years. Four males died, all from cancer. The estimated disease prevalence was 0.4/100,000 inhabitants. All the patients presented with anemia and polyps in the small bowel (80% in the duodenum, 66.7% in the ileum, and 40% in the jejunum), 93.3% underwent urgent surgical intervention and presented with intestinal invagination, and 40% of the patients developed cancer at a mean age of 48.5 years.
ConclusionThe present study is the first register of patients with Peutz-Jeghers syndrome in Valencia, Spain. The ICD-9 code is nonspecific for rare diseases. The duodenum was the most frequent location for polyps and the majority of cases presented with intestinal invagination, bowel obstruction, and urgent surgical intervention. A large percentage of patients presented with cancer. It would be of interest to review and evaluate the existing surveillance protocols in the Valencian Community.
El síndrome de Peutz Jeghers (SPJ) es una enfermedad rara con herencia autosómica dominante, causada por una mutación germinal del gen STK11/LKB1, localizado en el cromosoma 19p13.3, que consiste en hiperpigmentación mucocutánea, poliposis hamartomatosa y predisposición al cáncer. Se pretende identificar y registrar a los pacientes con SPJ, hacer una descripción de la enfermedad y estimar la prevalencia en Valencia (España).
Materiales y métodosSe obtuvo un listado de historias clínicas en 10 hospitales, utilizando el conjunto mínimo de base de datos de ingresos hospitalarios del Ministerio de Sanidad y Consumo de España utilizando el código CIE-9 759.6.
ResultadosSe encontraron 405 historias clínicas, de las que 15 pacientes estaban afectados (9 hombres y 6 mujeres), edad media de diagnóstico y fallecimiento de 13,8 y 54,2 años respectivamente, fallecieron 4 hombres, todos por cáncer; la prevalencia estimada es de 0,4/100.000 habitantes. Todos tenían anemia y pólipos en el intestino delgado (80% duodeno, 66,7% íleon y 40% en el yeyuno), el 93,3% tuvieron una intervención quirúrgica urgente y una invaginación intestinal, el 40% de pacientes desarrollaron cáncer con una edad media de 48,5 años.
ConclusiónEste es el primer estudio de registro de pacientes con SPJ en Valencia. La codificación CIE-9 es inespecífica para una enfermedad rara. La localización más frecuente de los pólipos fue el duodeno. La mayoría de casos ha tenido una invaginación intestinal, obstrucción intestinal e intervenciones quirúrgicas urgentes; un porcentaje elevado de pacientes presentó cáncer. Interesaría realizar un seguimiento y evaluación de los protocolos existentes en la Comunidad Valenciana.
Peutz-Jeghers syndrome (PJS) is characterized by the presence of hamartomatous polyposis of the gastrointestinal tract, mucocutaneous hyperpigmentation, and predisposition to cancer. It is a genetic disease that belongs to the group of rare diseases (RDs), which are defined in the European Union as diseases with a low prevalence of under 5 cases per 10,000 persons and a high mortality rate.1,2 It has an incidence peak in persons between 10 and 30 years of age.3,4 In 1895, the physician JT Connor was the first to describe the syndrome in 12-year-old identical female twins. They presented with pigmentation on the lips and in the mouth. One of them died at 20 years of age due to bowel obstruction, and the other died at 52 years of age from breast cancer.5 In 1921, the Dutch physician, Peutz, described 7 members of the Dutch Harrisburg family that presented with mucocutaneous pigmentation and polyposis in the intestine and nasopharynx. Two female family members had died before 20 years of age, due to bowel obstruction.6 In 1924, Van Dijk and Oudenal described two brothers with the characteristic mucocutaneous pigmentation in the mouth, intestinal polyposis, bowel obstruction, and numerous abdominal surgeries.7,8 Years later, Jeghers et al. (1949) described 10 cases from different families and published the characteristics of the syndrome.9 The eponym “Peutz-Jeghers syndrome” was introduced in 1954 by Bruwer et al., from the Mayo Clinic.5,10 The disease has broad genetic heterogeneity and variable and incomplete penetrance. The causal mutation is in the germline of the tumor suppressor STK11/LKB1 gene, located in the telomeric region of the short arm of chromosome 19p13.3,11 which encodes for a serine/threonine protein kinase (liver kinase B1 [LKB1]) that has several roles, depending on the molecular pathway involved. The relation between genotype and phenotype is not yet defined and the conclusion is that there is broad genetic homogeneity. 12–17 The presence of a mutation has been confirmed to cause loss of expression or function of STK11, a shortened protein or a truncated protein, with incomplete catalytic domains, resulting in the loss of kinase catalytic activity and disruption of the STK11 protein function.18,19 The role of STK11 is to act as a negative regulator (inhibitor) in various metabolic pathways and their alteration leads to decreased inhibition of cell development and growth, facilitating consequent uncontrolled cell growth, which explains the development of intestinal polyposis and tumors. The secondary alterations of the interaction between LKB1 and AMPK encompass different processes that finally produce effects on the function of STK11 in the suppression of cell growth and the regulation of energy homeostasis of the cell.
The clinical manifestations of PJS include hyperpigmented mucocutaneous lesions (85-99%)12,13,20 and polyposis that is generally gastrointestinal and hamartomatous, associated with the presentation of symptoms and complications, such as abdominal pain, gastrointestinal bleeding, secondary anemia, prolapse and ulceration of a polyp, bowel obstruction, and intestinal invagination. In the majority of cases, bowel obstruction and intestinal invagination require emergency laparotomies and bowel resections due to ischemia.14,21 Patients with the syndrome are also at an increased risk for developing gastrointestinal and extraintestinal cancers. Mucocutaneous lesion presentation is varied. They can be present at birth or appear in childhood and can disappear in adolescence. They are more frequently located in the oral mucosa (66-83%), either on the palate or gums (but not on the tongue),5 or the lips and perioral zone (94-96%). They can present around the eyes and nose (36%) and at the periocular level, with pigmentation on the eyelids and their edges and sometimes on the palpebral conjunctiva. They can also appear on the fingers, the dorsal and volar sides of the hands (74%) and feet (62%), and sometimes present in the perianal and genital regions and in the intestinal mucosa. The lesions are typically flat and can appear as scattered freckles or solitary greyish blue or purple spots, measuring approximately 1 to 5mm in diameter. The lesions rarely become malignant.5,10,16,17,20,22,23
Intestinal polyps are the next most frequent manifestation of PJS. Some patients only present with hyperpigmentation, whereas others present with both hyperpigmentation and intestinal polyps.24 They are found in the entire gastrointestinal tract, but more frequently in the small bowel (SB) (60-90%). The order of frequency is the jejunum, ileum, and duodenum, followed by the colon (50-63%), stomach (49%), and rectum (32%).17 They can also appear in other anatomic locations. They generally develop around early adolescence and are the cause of a large part of clinical morbidity. 25,26
Histologically, PJS polyps are hamartomatous and their morphology is characterized by the exaggerated and disorderly growth of the native cells of each organ from which they arise. Their features include disorganized “arboriform” growth of the smooth muscle (muscularis mucosae), perforating the lamina propria, and gland dilation within the submucosa or the muscularis propria.12 Growth is striking in SB polyps and less so in gastric polyps and colon polyps. SB and colon polyps tend to be pedunculated, whereas stomach polyps are sessile.12,25,27,28 The overlying epithelium of the polyp is typical of the bowel segment that is involved.
The increased risk for presenting with cancer includes gastrointestinal cancer (38-66%), breast cancer (32-54%), pancreatic cancer (11-36%), and less frequently thyroid cancer, lung cancer, and cancer of the reproductive organs (9-21%)3,27,29,30 The absolute risk for breast cancer has been described as similar to the risk observed in hereditary forms due to germline mutations of the BRCA1 or BRCA2 genes.29 Males can develop rare testicular tumors called calcifying Sertoli cell tumors that resemble sex cord tumors with annular tubules (SCTAT) and they can also develop gynecomastia and advanced bone age. The gynecologic findings associated with PJS include mucinous metaplasia of the Fallopian tube, endocervical adenocarcinoma, and ovarian tumors, such as cystic mucinous tumors and SCTAT tumors, which have been described in 4-year-old girls.25,27,31
The aim of the present study was to correctly identify and register the patients with PJS and estimate the prevalence of the disease in the province of Valencia, Spain, utilizing ICD-9 code 759.6 to search for patients. That code encompasses the unrelated diseases of PJS, von Hippel-Lindau syndrome (VHLS), Sturge-Weber syndrome (SWS), and other hamartoses not elsewhere classified in the Minimum Basic Data Set (CMBD, Spanish acronym) database. We shall describe some of the characteristics of the disease, provide an idea of the magnitude of the disease in our geographic region, and obtain the adequate registers. At present, no such instrument has been described or analysis published that uses said methodology to study PJS in Spain.
Materials and methodsStudy design, population, and instrumentsA retrospective, observational study of a case series from the province of Valencia, Spain, was conducted. According to data for 2016 from the National Statistics Institute (INE, Spanish acronym), the population was 2,517,951 inhabitants,32 which were the data used for the prevalence estimate. The present study was approved by the Clinical Research Ethics Committee of the Public Health Administration of Valencia and the ethics committees of 10 hospitals in the province of Valencia. The participating centers were the Hospital Universitario Doctor Peset Aleixandre, Hospital Universitario y Politécnico La Fe, Hospital General Universitario, Hospital Clínico Universitario, Hospital de Manises, Hospital de Sagunto, Hospital General de Requena, Hospital de la Ribera, Hospital Francesc de Borja de Gandia, Hospital Lluís Alcanyís de Xàtiva, and the Instituto Valenciano de Oncología.
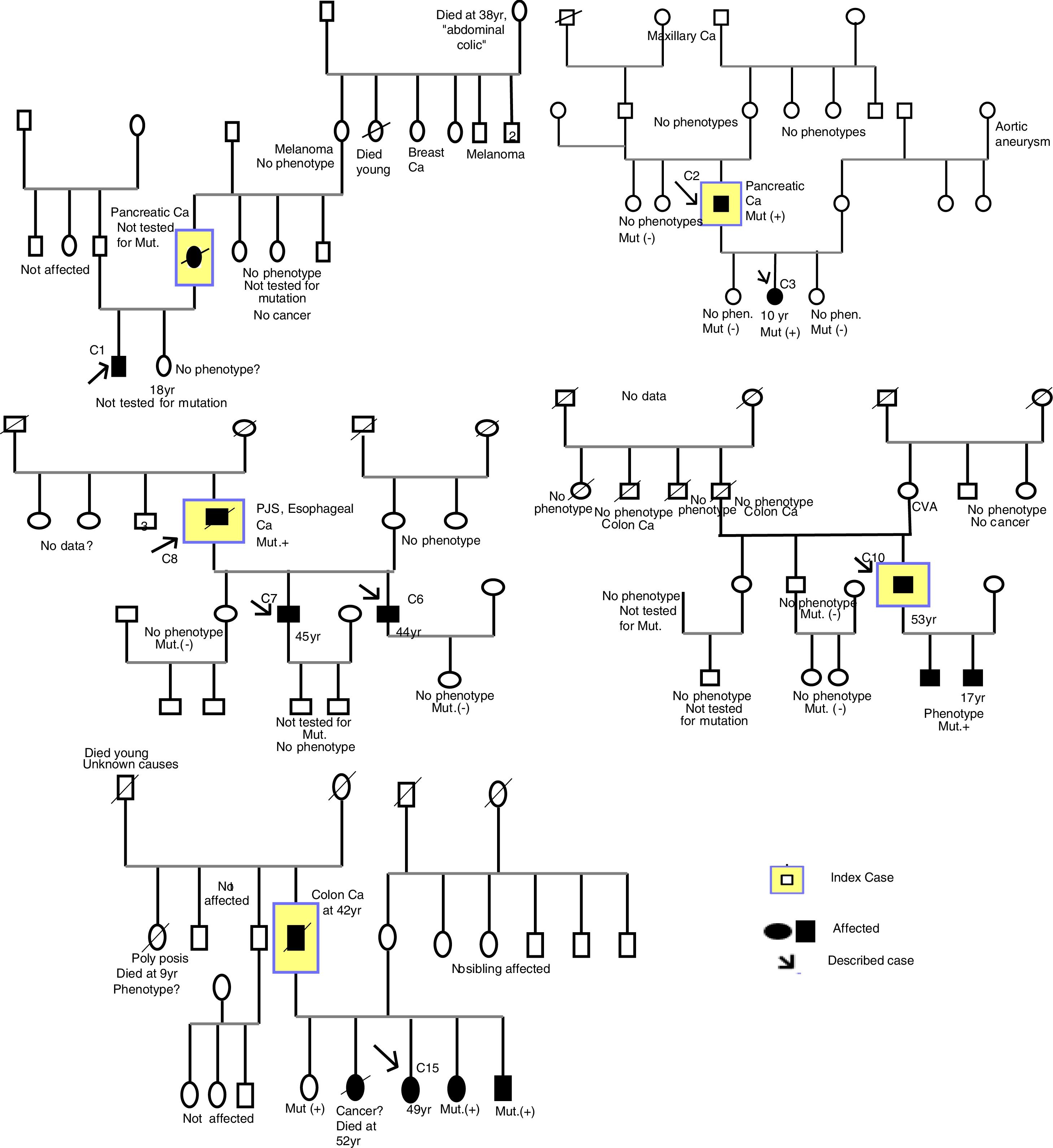
The procedure for identifying the patients was obtaining a print-out of the patient information registered in the CMBD database of hospital admissions of the Spanish Ministry of Health, Social Policy, and Equality, within the time frame of May 1, 1997 to May 1, 2016, utilizing code 759.6 of the ICD-9 classification system.33 From that list, each of the clinical histories (CHs) was reviewed to locate those with a diagnosis of PJS. In addition, a parallel search was carried out for patients diagnosed with PJS that were registered in the databases of the clinical computer software utilized at each hospital, with the term “Peutz-Jeghers”. The clinical software used for that search were Orion, Mizar, Iris, and Pangea. Each patient codified as PJS was contacted in person for their statements of informed consent. A thorough collection of the anonymized data and CH review was carried out, based on a questionnaire formulated for the present study that included different clinical and epidemiologic variables of the disease. Ten family trees were created (Fig. 1) and the cases included in the study were indicated. The index case was not always included, given that it did not necessarily come from a hospital in the province of Valencia or the patient had died.
: negative; (+): positive; Phen: phenotype; yr: years of age.")
To eliminate selection bias, after checking the cases of PJS with the initial lists, the inclusion of those patients in the database of the Promotion of Health and Prevention in the Healthcare Environment Service of the Public Health Administration, to which the Hereditary Cancer in the Valencian Community program belongs, was confirmed.
All personal data obtained for the present study were confidential and handled following the 2016/679 EU General Data Protection Regulation from April 27, 2016.
Inclusion and exclusion criteriaPatients were included in the study if they fit one of the following clinical criteria: 2 or more histologically confirmed Peutz-Jeghers (PJ)-type polyps, any number of PJ-type polyps in a patient with a family history of PJS in a close relative, characteristic mucocutaneous pigmentation in a patient with a family history of PJS in a close relative, and any number of PJ-type polyps in a patient with the characteristic mucocutaneous pigmentation.12,34,35
Patients registered with the ICD-9 code 759.6 that were not classified as PJS were excluded, i.e., the patients with diagnoses of SWS, VHLS, and other hamartoses (lung, breast, brain, gallbladder, soft tissue). Patients with “other diagnoses” that erroneously were codified as 759.6, such as angiomas, adenomas, Proteus syndrome, Cowden syndrome, Lhermitte-Duclos disease, and neurofibromatosis, were also excluded.
Description of variablesThe general variables of sex, current age, age at diagnosis, and age at death were collected. With respect to the clinical variables, they were considered if mucocutaneous pigmentation (melanosis) in the mouth, oral mucosa, nose, or surrounding areas, hands and fingers, feet and toes, and perianal region was present or not; if the patients had cancer, and if so, its location and patient age at cancer diagnosis; if the patient presented with polyps classified as hamartomatous, and if so, their location; and if the patient had another type of polyp, classified as adenomatous, hyperplastic, or mesenchymal.
Regarding the variables related to family history, a familial case was defined as a case in which 2 or more individuals in a family were affected; a positive family history (PFH) was a history that referred to the presence of cancer related to PJS, to a relative with PJS, and to the characteristic mucocutaneous pigmentation and/or polyposis in a first-degree relative; and a sporadic case was defined as a case in which the clinical or genetic diagnosis was made in only one family member, as long as the clinical, endoscopic, and radiologic presence of hamartomas and mucocutaneous pigmentation was ruled out in the parents and/or if the mutation was known, if it was absent, and if the case did not fit the PFH criteria.
The variables related to the associated complications were: having undergone a first non-endoscopic, invasive emergency gastrointestinal surgery during the course of the disease and age at the time of PJS diagnosis, bowel obstruction, intestinal invagination and age at presentation, rectal bleeding, a prolapsed polyp, an ulcerated polyp, anemia, upper gastrointestinal bleeding (UGIB), bowel perforation, and finally, the number of hospital admissions throughout the course of the disease.
Another variable was whether the patient had received genetic counseling at a Hereditary Cancer Genetic Counseling Unit (HCGCU) at a first visit and/or continued control and follow-up at the unit within the past 10 years.
The genetic test variable, in relation to whether or not testing for the STK11 gene mutation was performed, its result, and the description of the mutation, was also described.
Genetic testTo obtain the genetic analysis test results, the different genetic services and those of genetic counseling on hereditary cancer in Valencia (Hospital Universitario y Politécnico La Fe, Hospital Universitario Clínico de Valencia, and Instituto Valenciano de Oncología) were contacted. The majority of the patients were referred to the Molecular Genetics Unit of the Hospital General Universitario de Elche, which is where the genetic tests of the patients in the Valencian Community Hereditary Cancer program36 are performed. One test was carried out at the Molecular Biology Laboratory of the Hospital Universitario y Politécnico La Fe, because at the time that test was performed, samples were not yet being referred to the Hospital de Elche.
Statistical analysisThe data were captured in an IBM SPSS version 24.0 software database. The descriptive analysis utilized mean and standard deviation. The Levene’s test and Mann-Whitney U test were used to compare the variances and means of the 2 independent groups with normal and abnormal distribution, respectively. The categorical variables were described using frequencies and percentages. Family trees were created utilizing the GenoPro 2018 program.
ResultsCollection and construction of the patient registerA print-out of 405 clinical histories codified as ICD-9 759.6 was obtained. Twenty-two patients were assigned the PJS diagnosis, but only 15 cases fit the diagnostic criteria. Of the cases that did not meet the diagnostic criteria, one was a codification error, given that the patient had no symptoms for suspecting PJS, one case was a deceased newborn whose CH stated a family history of PJS (it was not possible to locate any of the relatives), 2 cases were suspected of uninvestigated PJS, and 3 cases were suspected of investigated PJS and ruled out. C1 belonged to a different autonomous community and C3 and C11 belonged to another province of the Valencian Community but they were included in the study because they had received hospital care and appeared in the hospital admissions register of one of the hospitals of the province of Valencia (Hospital Universitario y Politécnico La Fe).
Description of cases and families, family history, and defined familial casesEleven families (Fs) and 15 cases (Cs) were described. There was 1 case for each family in F5, F6, F7, F8, F9, and F10 and 2 cases for each family in F2 and F3 (F3 refused to have their family tree created). There were 5 affected members in F11 (father, 3 sisters, and one brother), but only one case was included (sister, C15); there were 2 cases in F1 (mother and son), but only one case was included (son, C1); and there were 3 affected members in F10 (father and 2 sons), but only one case was included (father, C10). Some cases were excluded because they had never sought treatment at a hospital in the province of Valencia, their CH data could not be accessed, or they did not appear in the hospital admissions register of the hospitals in the province of Valencia (Fig. 1).
All the cases were natives of Spain, except the F4 cases, who were natives of Bulgaria.
To classify a case as a defined familial case and as a case with a PFH, we analyzed the family trees and the clinical data collected. From the CH data, C4 and C5 (F3) were documented as defined familial cases. Ten (67%) of the 15 cases were defined familial cases and were made up of 6 families. Only 6 (60%) cases of the 10 had a PFH (C1, C3, C5, C6, C7, C15). The family history was unknown in 2 cases and the other 2 cases did not have a PFH. Six of the 15 cases had a PFH, 6/15 (40%) cases were sporadic cases, and 3 cases could not be defined (C4, C8, C12) due to unknown histories (Table 1).
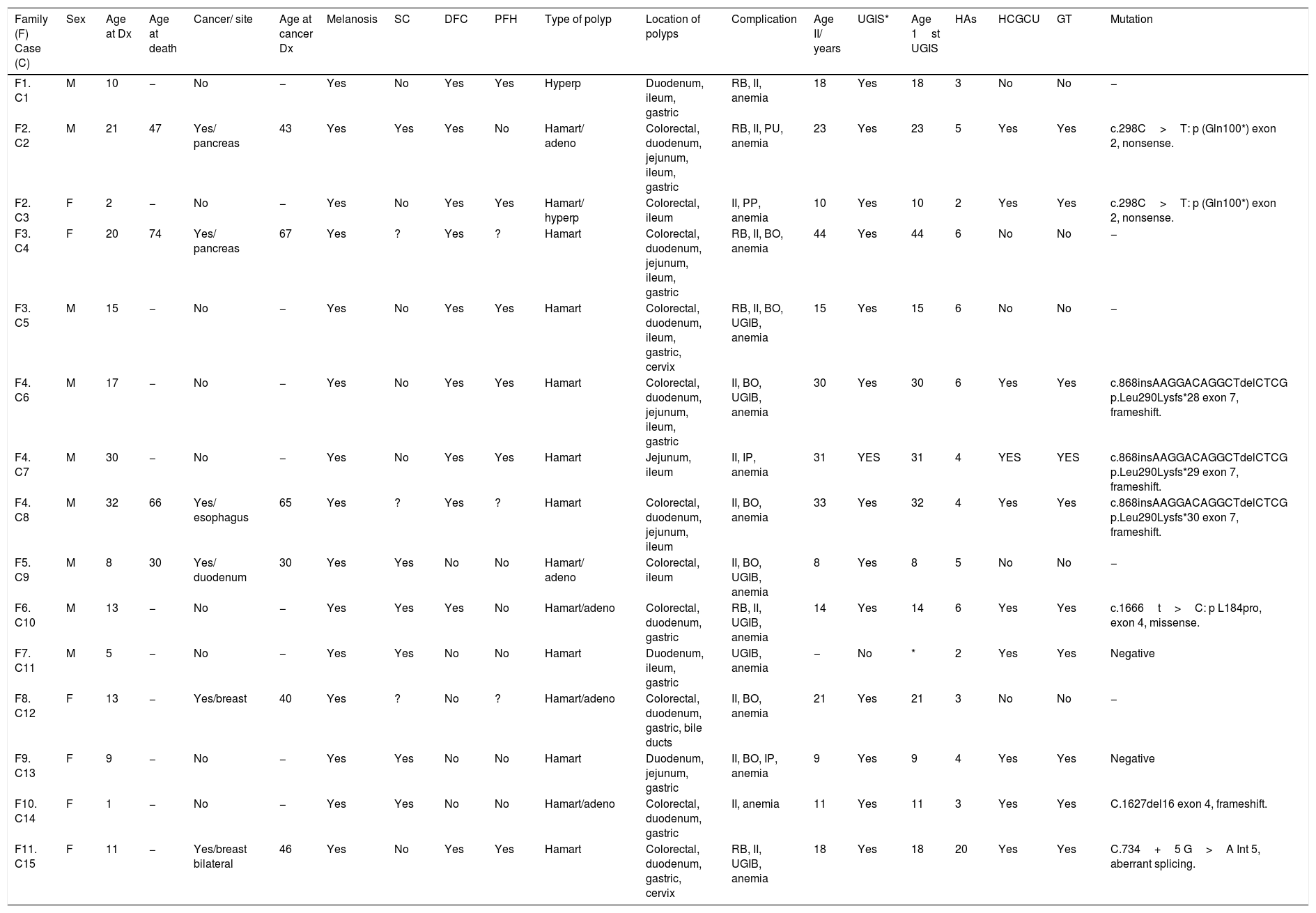
Case descriptions.
| Family (F) Case (C) | Sex | Age at Dx | Age at death | Cancer/ site | Age at cancer Dx | Melanosis | SC | DFC | PFH | Type of polyp | Location of polyps | Complication | Age II/ years | UGIS* | Age 1st UGIS | HAs | HCGCU | GT | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1. C1 | M | 10 | − | No | − | Yes | No | Yes | Yes | Hyperp | Duodenum, ileum, gastric | RB, II, anemia | 18 | Yes | 18 | 3 | No | No | − |
| F2. C2 | M | 21 | 47 | Yes/ pancreas | 43 | Yes | Yes | Yes | No | Hamart/ adeno | Colorectal, duodenum, jejunum, ileum, gastric | RB, II, PU, anemia | 23 | Yes | 23 | 5 | Yes | Yes | c.298C>T: p (Gln100*) exon 2, nonsense. |
| F2. C3 | F | 2 | − | No | − | Yes | No | Yes | Yes | Hamart/ hyperp | Colorectal, ileum | II, PP, anemia | 10 | Yes | 10 | 2 | Yes | Yes | c.298C>T: p (Gln100*) exon 2, nonsense. |
| F3. C4 | F | 20 | 74 | Yes/ pancreas | 67 | Yes | ? | Yes | ? | Hamart | Colorectal, duodenum, jejunum, ileum, gastric | RB, II, BO, anemia | 44 | Yes | 44 | 6 | No | No | − |
| F3. C5 | M | 15 | − | No | − | Yes | No | Yes | Yes | Hamart | Colorectal, duodenum, ileum, gastric, cervix | RB, II, BO, UGIB, anemia | 15 | Yes | 15 | 6 | No | No | − |
| F4. C6 | M | 17 | − | No | − | Yes | No | Yes | Yes | Hamart | Colorectal, duodenum, jejunum, ileum, gastric | II, BO, UGIB, anemia | 30 | Yes | 30 | 6 | Yes | Yes | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*28 exon 7, frameshift. |
| F4. C7 | M | 30 | − | No | − | Yes | No | Yes | Yes | Hamart | Jejunum, ileum | II, IP, anemia | 31 | YES | 31 | 4 | YES | YES | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*29 exon 7, frameshift. |
| F4. C8 | M | 32 | 66 | Yes/ esophagus | 65 | Yes | ? | Yes | ? | Hamart | Colorectal, duodenum, jejunum, ileum | II, BO, anemia | 33 | Yes | 32 | 4 | Yes | Yes | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*30 exon 7, frameshift. |
| F5. C9 | M | 8 | 30 | Yes/ duodenum | 30 | Yes | Yes | No | No | Hamart/ adeno | Colorectal, ileum | II, BO, UGIB, anemia | 8 | Yes | 8 | 5 | No | No | − |
| F6. C10 | M | 13 | − | No | − | Yes | Yes | Yes | No | Hamart/adeno | Colorectal, duodenum, gastric | RB, II, UGIB, anemia | 14 | Yes | 14 | 6 | Yes | Yes | c.1666t>C: p L184pro, exon 4, missense. |
| F7. C11 | M | 5 | − | No | − | Yes | Yes | No | No | Hamart | Duodenum, ileum, gastric | UGIB, anemia | − | No | * | 2 | Yes | Yes | Negative |
| F8. C12 | F | 13 | − | Yes/breast | 40 | Yes | ? | No | ? | Hamart/adeno | Colorectal, duodenum, gastric, bile ducts | II, BO, anemia | 21 | Yes | 21 | 3 | No | No | − |
| F9. C13 | F | 9 | − | No | − | Yes | Yes | No | No | Hamart | Duodenum, jejunum, gastric | II, BO, IP, anemia | 9 | Yes | 9 | 4 | Yes | Yes | Negative |
| F10. C14 | F | 1 | − | No | − | Yes | Yes | No | No | Hamart/adeno | Colorectal, duodenum, gastric | II, anemia | 11 | Yes | 11 | 3 | Yes | Yes | C.1627del16 exon 4, frameshift. |
| F11. C15 | F | 11 | − | Yes/breast bilateral | 46 | Yes | No | Yes | Yes | Hamart | Colorectal, duodenum, gastric, cervix | RB, II, UGIB, anemia | 18 | Yes | 18 | 20 | Yes | Yes | C.734+5 G>A Int 5, aberrant splicing. |
ADC: adenocarcinoma; Adeno: adenomatous; BO: bowel obstruction; DFC: defined familial case; Dx: diagnosis; F: female; GI: gastrointestinal; GT: genetic test; HAs: hospital admissions; Hamart: hamartomatous; HCGCU: Hereditary Cancer Genetic Counseling Unit; Hyperp: hyperplastic; II: intestinal invagination; IP: intestinal perforation; M: male; PFH: positive family history; PP: prolapsed polyp; RB: rectal bleeding; SC: sporadic case; UGIB: upper gastrointestinal bleeding; UGIS: urgent gastrointestinal surgery; UP: ulcerated polyp;? : unknown.
Seven of the 11 families and 10 of the 15 cases underwent genetic testing. The mutation was identified in 5 families (71%) and 8 cases (80%). C10 initially presented with a variant with an unknown clinical effect or significance, but according to the database revisions, could currently be considered to have a likely pathogenic variant. Only C11 and C13 (20%) were negative.
Genetic testing was performed on 4 of the 6 cases with a PFH. A pathogenic mutation was identified in all of them. Genetic testing was not requested for C1, and C5 did not wish to be tested.
Table 1 shows the mutations found. There were 5 mutations: one (20%) nonsense (F2), one (20%) missense (F6), 2 (40%) frameshift (F4 and F10), and one (20%) aberrant splicing (F11).
Epidemiologic characteristicsOf the 15 cases, 9 were males (60%) and 6 were females (40%), with a male:female ratio of 1.5:1. The age range of the participants was from 10 to 60 years. The estimated actual prevalence for the year 2016 was 0.4/100,000 inhabitants.
The range of age at diagnosis was from one to 32 years, with a mean age at diagnosis of 13.8±9.08 years. For males, it was 17.3±9.40 years and for females it was 8.5±5.78 years (p=0.062). The mean age at death was 54.2±19.7 years.
Clinical presentation and complicationsAll the patients presented with the mucocutaneous pigmentation phenotype and had polyps. The polyps in C1 were hyperplastic, not hamartomatous, but fit the criteria for PJS. In addition, 5 cases presented with adenomatous polyps, diagnosed in the patients, between 13 and 43 years of age (mean age was 31±15.5 years), after the diagnosis of PJS. Importantly, the cases with adenomatous polyps, C2 and C9, presented with pancreatic cancer and SB (duodenal) cancer, respectively. Only C3 presented with one hyperplastic polyp 7 years after diagnosis and did not present with any type of cancer.
Regarding polyp location, all the cases had polyps in the SB (80% in the duodenum, 67% in the ileum, and 40% in the jejunum). Eleven cases (73.3%) presented with colorectal polyps and 11 (73.3%) with gastric polyps. We emphasize that all the cases had polyps in more than one location. Three cases (20%) presented with extraintestinal polyps (C12 in the gallbladder and C5 and C15 in the uterine cervix).
All the cases had suffered with a gastrointestinal complication. Fourteen (93.3%) had presented with an intestinal invagination (at a mean age of 19±10.3 years), 9 (60%) with bowel obstruction (at a mean age of 25±11.5 years), 6 (40%) with rectal bleeding (at a mean age of 26±12.3 years), 6 (40%) with UGIB (at a mean age of 23±9.7 years), one (6.7%) with a prolapsed polyp (at 2 years of age), and one (6.7%) with an ulcerated polyp (at 30 years of age). All the cases had presented with anemia. Fourteen cases (93.3%) had required a first emergency gastrointestinal surgery (at a mean age of 19±10.2 years) and a mean number of hospital admissions of 5±4.3 throughout the course of the disease.
Presentation of cancerOf all the patients, 6 cases (40%) presented with cancer, with a mean age at cancer diagnosis of 48.5±14.60 years (range of 30 to 67 years). Four (67%) of those patients were males and 2 (33%) were females. According to age at cancer diagnosis, C9 presented with cancer the earliest. Cancer was the cause of death in the 4 patients that died. Thirteen percent of the cases presented with pancreatic cancer, at a mean age of 55 years. Of the 8 cases with the STK11 mutation, 3 developed cancer (37.5%). Breast cancer and pancreatic cancer were the most frequent. Table 1 describes the characteristics, histologic type of cancer, and age at diagnosis.
Genetic counseling on hereditary cancerTen cases (67%) sought attention at an HCGCU. Five cases (33%) did not: C4 and C9 died more than 10 years earlier, C1 was not referred, the reason was unknown for C5, and C12 did not wish to.
DiscussionIn some countries, data from the registers of patients with PJS are available, thanks to national polyposis registers.37 One of the oldest polyposis registers in the world is the St. Mark’s Hospital Register in London.16 Tan et al. (2010) conducted a study in Singapore,26 and from 610 cases in the polyposis register, identified only 7 cases of PJS. No polyposis register has been reported in Spain, but there is a specific familial adenomatous polyposis register.38
As in the present analysis, in the study by Fostira et al. (2018) on 8 Greek patients and the study by Tchekmedyian et al. (2013) on 25 Uruguayan patients, those authors described the creation of a register of patients with PJS.30,39 There are academic articles on retrospective, observational studies conducted worldwide that utilize different methodologies to identify the patients. Some use clinical laboratory databases and the genetic test for STK11, and others use histopathologic laboratory databases related to hamartomatous polyposis or cancer, but there is no precise methodology described for obtaining an adequate patient register.14,25,38
PJS requires an accurate and universal nomenclature and classification. Different classifications have been created. For example, the database of RDs and orphan drugs, Orphanet,40,41 classifies PJS with the number 2869, but within several subcategories and groups. Other codifications are included in that database, such as the one described by Online Mendelian Inheritance in Man®. PJS is assigned #175200 and the focus is on the relation between phenotype and genotype. The MeSH code D010580 and the MedDRA code 10034764, which are normalized medical terminology for exchanging information, are also included. On the other hand, anatomopathologists created an international nomenclature system of restricted use called SNOMED-CT. That system was used with code S54320 by Jelsig et al. (2016) to identify the cases in their study on 46 Danish patients.20
For more than 20 years, the ICD-9 coding system33 has been used in Spain and it includes approximately 100 codes for RDs. The European Union recommended the development of an RD inventory to be implemented in the new ICD-10 coding system.42 The ICD-10 classification includes 300 codes for RDs, providing more possibilities for cataloguing and classifying an RD, thus facilitating the comprehensive study of RDs related to each other.1 However, in the case of PJS, the ICD-10 code is Q85.8, described as “other phakomatoses not elsewhere classified”, and as in the ICD-9 code, it also groups PJS, SWS, and VHLS together, which are diseases that are not related to each other. At least the diagnosis of “other hamartoses not elsewhere classified” was excluded, thus eliminating from the search lists with large numbers of patients erroneously coded and included within a general classification such as “other hamartoses”, as well as endless diagnoses of hamartomas in different anatomic sites. Other RDs with a different specific code can also be ruled out. A conversion of all ICD-9 codes to ICD-10 codes is currently being carried out in Spain, and databases and registers of RDs are also being created in an effort to universalize the common use of the Online Mendelian Inheritance in Man®, Orpha, and ICD-10 codes.
In the present study, we used the ICD-9 code because it was still being used during the study period and the data were not completely reclassified and converted to ICD-10 at the time of their collection. Even though the ICD-9 classification is not very specific, a clinical description of the cases in the province of Valencia was found, validated, and carried out.
In our methodology, we used the CMBD database because many of our patients were seen at tertiary care hospitals, needing emergency service care, and on some occasions required hospital admission, either for severe complications, complementary tests, or special in-hospital treatments.1 In conducting the present study, we found that the validation and follow-up of patients with PJS required the review of a large number of CHs that resulted in an n of only 15 patients. Perhaps the low prevalence of the RD justified the low n of the study, but we cannot ignore the fact that we did not have a more accurate way to screen and register the cases of PJS because a more specific coding and classification system was not available to us at the time of the analysis. The classification system we used made the epidemiologic searches for and validations of RDs very time-consuming, and given the small number of cases identified, not very successful. Nevertheless, the ICD-9 classification appears to be used for searches of cases in other parts of the world. For instance, a study conducted in the United States on children utilized the ICD-9 classification as a search tool.43 Interestingly, based on the Danish National Patient Register, Jelsig et al. utilized the ICD-10 code DQ858B, but in that register they used extensions and added A, B, and C to make codes for specific diseases. For example, in the case of PJS a B was added to the international ICD-10 code, thus making the register of an RD more specific.20,44
Possible selection bias could be a significant limitation of the present study. Because the analysis is a retrospective review that included patients that sought medical attention at a hospital, asymptomatic children might not have appeared on that register, and thus were not included in the study.
The reported prevalence figures for the disease vary and bibliographic reviews show that different methodologies for its calculation are used. Prevalence from 1:50,000 to 1:280,000 are reported in numerous review articles.20,30,45–47 Orphanet’s second report for 2017 describes an estimated prevalence in Europe of 0.4 per 100,000 births.48 Reports of incidence range from 1:8,300 to 1:200,000 births.3,12,13,18,35,39,49 The estimated prevalence in our case series was 0.4/100,000 inhabitants for 2016, which is within the ranges described in other publications.
Mean ages at diagnosis have been reported from 17 to 26 years of age.17,26,39,50–52 The mean age at diagnosis of the present study remained within a pediatric age, and age in males was almost double that of females, but with no statistically significant differences. No predominance in either sex5,46,47 or in any race17 has been described. In a Uruguayan study a male:female ratio of 1:0.6739 was reported, concurring with our results but differing from the 1:2.5 described by Tan et al.26
The mean age at the time of death in the present case series was 5 to 10 years greater than the 49.5 years reported in the Danish study and the 44 years reported in the Uruguayan study.20,39 In our study, the most frequent cause of death was cancer, coinciding with the results of the Uruguayan study, and the mean age at death of those patients was also similar.39 Given the above information, patients with PJS appear to die within a young age range.
In 1996, Tomlinson et al. initially ruled out probable loci on chromosomes 1 and 6. 4 Later, Hemminki et al. (1998)11 described the causal mutation in the germline of the STK11 gene, which has a length of 23 Kb and contains 9 exons that encode for a serine-threonine protein kinase with 433 amino acids (LKB1) and a final non-coding exon. The coding or catalytic region (protein kinase domain) is found between amino acids 44 and 309. The C-terminal and N-terminal regions are not analogous to other kinases of the same family. The LKB1 protein is expressed in all human tissue, but with greater intensity in the fetal testes and liver, and it is found in the nucleus and cytoplasm of the cell. Another probable locus on 19q13 has been suggested.13,53,54
There are more than 300 human STK11 gene mutations in the NCBI database (https://www.ncbi.nlm.nih.gov/clinvar).55 Approximately 85% are small mutations, and of those, 35% are point mutations that include the missense and nonsense types, 50% are small insertions and deletions, and 15% affect the splicing sites,56,57 concurring with our results of 40%, 40%, and 20%, respectively. The 5 types of mutations found in our study are currently described in databases.
There can be a mutation in 50-94% of affected cases.8,12,25,30,50,58 A high percentage of cases (80%) and families (71%) in our study that underwent gene testing had a mutation, coinciding with the results of the Danish analysis and the study by Fostira et al. that reported mutations in 86% and 87.5% of the patients, respectively. In contrast, Scott et al. (2002) reported a mutation in less than 50% of the 14 Australian patients studied, and in a case series of 51 patients in the United States, Amos et al. (2004) described a mutation in 69% of the patients tested.51,59 An LKB1 gene mutation was previously thought to be found in the majority of patients, but McGarrity et al. (2000) proposed that not all cases of PJS were caused by a mutation in said gene, given that they identified it in 60% of the familial cases and 50% of the sporadic cases. That could be explained by the use of different mutational analysis methods, the possibility of heterogeneity of the locus and the disease, or because there really is a second gene responsible for PJS.
Twenty-five to 30% of cases PJS are reported to be sporadic, possibly due to de novo mutations or to variants with low penetrance.6,24 Mutations have been identified in 50-90% of sporadic cases.13,28,51 In our case series, the sporadic case percentage was 10-15% higher than that described (Table 1), which could be because the definition of sporadic case in the majority of studies includes genetic testing of the case and the parents, whose results must be negative.13,50 We did not include the condition of genetic testing for defining a case as sporadic, because in the past, our population did not have the current access to said testing, and before 2008, no protocol for PJS was included in the hereditary cancer programs within the Valencia healthcare system.
Numerous studies report that 36-50% of the cases have a PFH, coinciding with the results of our case series20,22,26,30,39,51 (Table 1).
Several studies have shown that hyperpigmented macules are the most frequent and typical clinical characteristics of the pathology, coinciding with our study findings.13,26,39,50
Eighty to 100% of the patients present with gastrointestinal polyposis. All of our patients had polyps, including the hyperplastic and adenomatous types. The same was documented in the Danish study.20 Of the cases in our study that presented with a second adenomatous polyp, one developed cancer of the SB, which could be associated with the possible transformation of a portion of hamartomatous polyps into adenomatous polyps and then into carcinoma that is described in the literature.22,60,61 Bardeesy et al. (2002) suggested that the loss of STK11 gene expression in the epithelium resulted in benign polyps and that the loss of expression in a late-stage lesion facilitated the development of potentially malignant lesions.62
Polyps can appear at other sites, such as the nasal cavities, bronchi, biliary tract, ureter and bladder, uterine cervix, and vagina.12,20,25,27,39,63 Three of our patients presented with extraintestinal polyps.
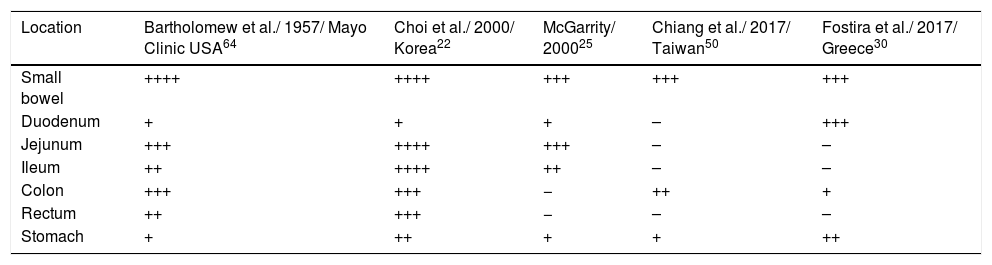
Polyps occur more frequently in the SB (64-96%).26,39 All the patients in our case series presented with polyps in the SB, followed by the stomach and colon, concurring with that reported in the Taiwanese study.50 In Table 2, the locations of polyps described as the most frequent are summarized. In the SB, polyps are most frequently found in the jejunum, but they were more frequent in the duodenum in our study.22,25,26,64
Most frequent polyp locations in patients with PJS.
| Location | Bartholomew et al./ 1957/ Mayo Clinic USA64 | Choi et al./ 2000/ Korea22 | McGarrity/ 200025 | Chiang et al./ 2017/ Taiwan50 | Fostira et al./ 2017/ Greece30 |
|---|---|---|---|---|---|
| Small bowel | ++++ | ++++ | +++ | +++ | +++ |
| Duodenum | + | + | + | – | +++ |
| Jejunum | +++ | ++++ | +++ | – | – |
| Ileum | ++ | ++++ | ++ | – | – |
| Colon | +++ | +++ | − | ++ | + |
| Rectum | ++ | +++ | − | – | – |
| Stomach | + | ++ | + | + | ++ |
++++: very frequent; -: not reported.
The most frequent complications associated with polyps are generally surgical and urgent. Approximately 50% of patients present with abdominal symptoms before 20 years of age, particularly secondary to bowel obstruction and intussusception.17 Invaginations are generally caused by polyps>de 15mm and treatment is principally surgical.65 Patients with PJS are at a high accumulated risk for presenting with intestinal invagination at an early age. In their case series on 110 Dutch patients, Van Lier et al. (2011) reported a risk for intestinal invagination of 15% (95% CI ± 8-22%) at 10 years of age, 50% (± 40-60%) at 20 years of age, 65% (± 55-75%) at 30 years of age, 77% (± 68-86%) at 40 years of age, and 84% (± 75-93%) at 50 years of age.65 In our study, anemia and intestinal invagination were the most frequent complications, followed by bowel obstruction, rectal bleeding, UGIB, a prolapsed polyp, and an ulcerated polyp. The study by Choi et al. (2000) on 30 Korean patients and the Taiwanese, Uruguayan, and Dutch studies all reported that invagination was one of the most frequent complications and the major cause of emergency abdominal surgery, coinciding with our results. The reported mean ages of patients at the presentation of an invagination is 16 to 20 years, and in our patients, it was 19 years.22,39,50,65 The authors of a case series reported that the median age at the appearance of intussusception was 10 years, and 40% of the 14 pediatric patients with PJS had already developed the complications of intestinal invagination and rectal bleeding at 8 years of age.43 We emphasize the fact that in our cases series, the majority of the cases required a first upper gastrointestinal emergency surgery at a mean age of 19 years and presented with complications and surgical interventions at an early age. That could be due to an intrinsic or extrinsic deficiency in the control and/or follow-up of the polyps, because they are the main cause of invaginations.
Anemia was a cause of significant morbidity, given that all our patients presented with it, but it could not be related to a specific etiology, even though 40% of the patients had rectal bleeding and 40% had upper gastrointestinal bleeding. Microscopic intestinal bleeding could not be ruled out in the other patients because the CHs did not reliably show that serial fecal occult blood testing had been carried out. Tan et al.26 associated gastrointestinal bleeding with anemia and stated that it conferred morbidity on 42% (n=3) of the cases. Lower than our results, the Uruguayan study reported that only half of the patients presented with anemia.39
No studies were found in the literature that mentioned the number of hospital admissions for patients with PJS and so we could not make that comparison with our results.
Table 3 describes the most frequent sites of cancer in patients with PJS.3,5,39,52,66,67 In our case series, the most frequent tumors were in the breast and pancreas. Non-gastrointestinal cancer sites have been described in the breast, endometrium, cervix, ovary, lung, gallbladder,13 thyroid,20,39 nasal cavity or sinus, penis, kidney, and prostate.50,68,69 Other types of lesions, such as SCTAT in the ovary, calcifying Sertoli cell tumors of the testes, and gonadal stromal testicular tumors have been described, with a mean patient age at the time of presentation of testicular cancer of 9 years (95% CI: 4-13) and a range from 3 to 20 years.29 Those types of tumors were not diagnosed in our case series.
Tumor appearance frequency in patients with PJS.
| Hearle et al./ 2006/ EU, Australia, USA66 | Van Lier M. et al./ 20103 | Resta et al./ 2013/ Italy52 | Tchekmedyan et al./ 2013/ Uruguay39 | Ishida et al./ 2016/ Japan67 | |
|---|---|---|---|---|---|
| GIT | ++++ | − | +++ | − | − |
| Gastroesophageal | ++ | − | − | − | + |
| Stomach | − | ++ | − | − | ++++ |
| Small bowel | ++ | +++ | − | + | ++ (duodenum) |
| Jejunoileal | − | − | − | − | ++++ |
| Colorectal | +++ | +++++ | − | − | +++++ |
| Pancreas | ++ | + | − | − | +++ |
| Extraintestinal | |||||
| Breast | +++ | ++++ | ++ | +++ | ++ |
| Thyroid | − | − | − | ++ | + |
| Lung | + | − | − | + | ++ |
| Bladder | − | − | − | − | ++ |
| Kidney | − | − | − | + | + |
| Uterus | − | − | − | − | ++++++ |
| Gynecologic | + | − | − | − | ++ |
+++++: very frequent, -: not reported; EU: European Union; GIT: gastrointestinal tract (colon, small bowel, esophagus, stomach, and pancreas); Gynecologic: uterus, ovary, cervix; USA: United States of America.
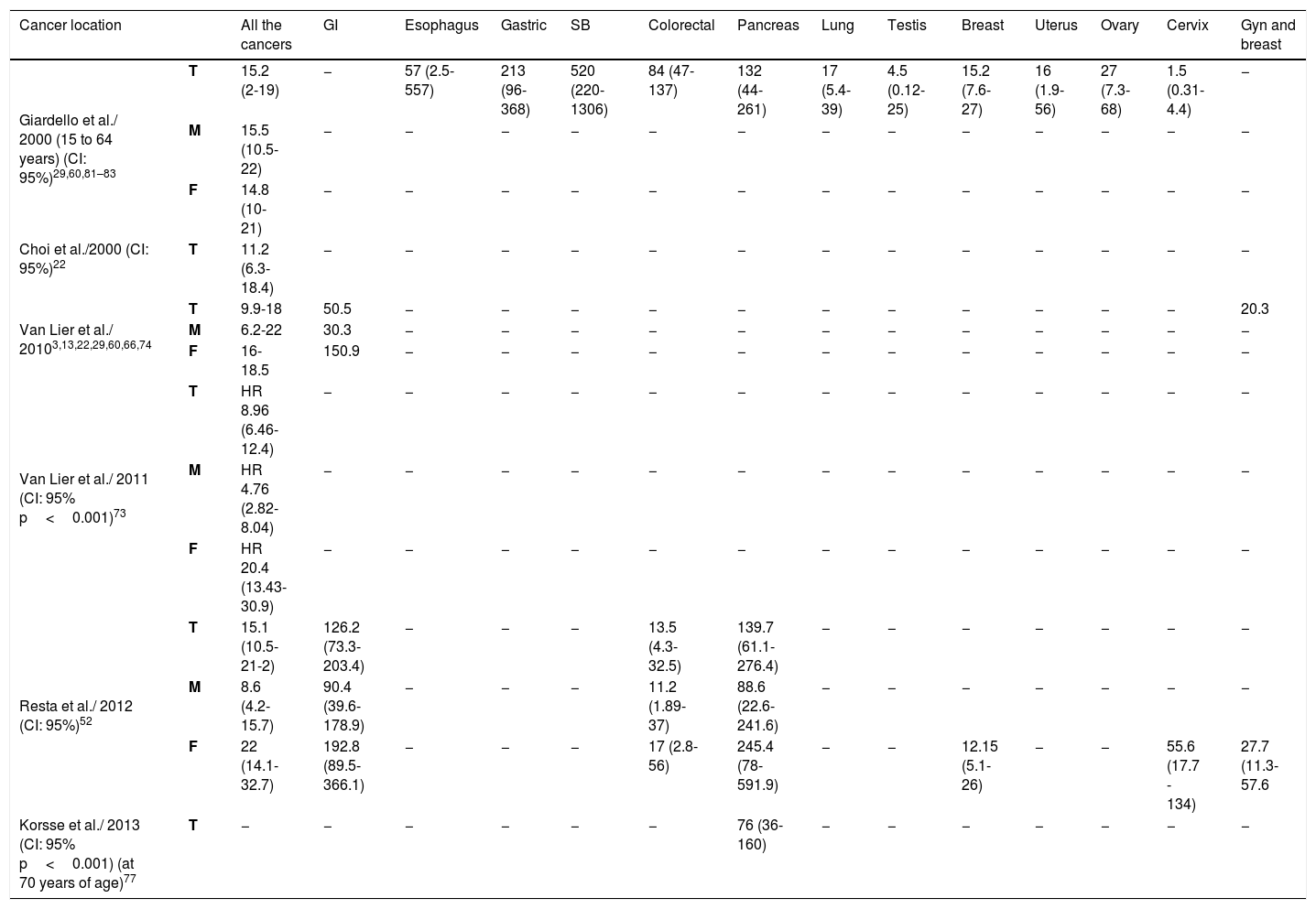
In addition to control and surveillance of polyps, another goal of the follow-up protocols for PJS is screening for and early detection of cancer, given that cases of cancer have been described from the age of 60 years in patients that suffer from PJS.61,70–72 Numerous studies using different methodologies have been published whose aim has been to show that patients with PJS have a high risk for presenting with gastrointestinal or extraintestinal cancer, reporting the accumulated risks (ARs) and relative risks (RRs) for developing cancer.60Table 4 compares the different RRs described. All the authors coincide in describing a RR from 9.9 to 18 for any type of cancer. In a meta-analysis, Giardello et al. (2000) also observed a statistically significant increase in the RR for cancer of the esophagus, stomach, SB, colon, pancreas, lung, breast, uterus, and ovary and Resta et al. found a high relative risk for gastrointestinal and gynecologic cancers in women, particularly for pancreatic cancer and cervical cancer. All the authors reported higher RRs in women than in men, whereas our study found more cancer diagnoses in men than in women.3,22,29,52,73
Relative risk of cancer reported in patients with PJS.
| Cancer location | All the cancers | GI | Esophagus | Gastric | SB | Colorectal | Pancreas | Lung | Testis | Breast | Uterus | Ovary | Cervix | Gyn and breast | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Giardello et al./ 2000 (15 to 64 years) (CI: 95%)29,60,81–83 | T | 15.2 (2-19) | − | 57 (2.5-557) | 213 (96-368) | 520 (220-1306) | 84 (47-137) | 132 (44-261) | 17 (5.4-39) | 4.5 (0.12-25) | 15.2 (7.6-27) | 16 (1.9-56) | 27 (7.3-68) | 1.5 (0.31-4.4) | − |
| M | 15.5 (10.5-22) | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| F | 14.8 (10-21) | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Choi et al./2000 (CI: 95%)22 | T | 11.2 (6.3-18.4) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Van Lier et al./ 20103,13,22,29,60,66,74 | T | 9.9-18 | 50.5 | − | − | − | − | − | − | − | − | − | − | − | 20.3 |
| M | 6.2-22 | 30.3 | − | − | − | − | − | − | − | − | − | − | − | − | |
| F | 16-18.5 | 150.9 | − | − | − | − | − | − | − | − | − | − | − | − | |
| Van Lier et al./ 2011 (CI: 95% p<0.001)73 | T | HR 8.96 (6.46-12.4) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| M | HR 4.76 (2.82-8.04) | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| F | HR 20.4 (13.43-30.9) | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| Resta et al./ 2012 (CI: 95%)52 | T | 15.1 (10.5-21-2) | 126.2 (73.3-203.4) | − | − | − | 13.5 (4.3-32.5) | 139.7 (61.1-276.4) | − | − | − | − | − | − | − |
| M | 8.6 (4.2-15.7) | 90.4 (39.6-178.9) | − | − | − | 11.2 (1.89-37) | 88.6 (22.6-241.6) | − | − | − | − | − | − | − | |
| F | 22 (14.1-32.7) | 192.8 (89.5-366.1) | − | − | − | 17 (2.8-56) | 245.4 (78-591.9) | − | − | 12.15 (5.1-26) | − | − | 55.6 (17.7 - 134) | 27.7 (11.3-57.6 | |
| Korsse et al./ 2013 (CI: 95% p<0.001) (at 70 years of age)77 | T | − | − | − | − | − | − | 76 (36-160) | − | − | − | − | − | − | − |
CI: confidence interval; F: female; GI: gastrointestinal (colorectal, small bowel, gastric, esophagus, pancreas); Gyn: gynecologic; M: male; SB: small bowel; T: total.
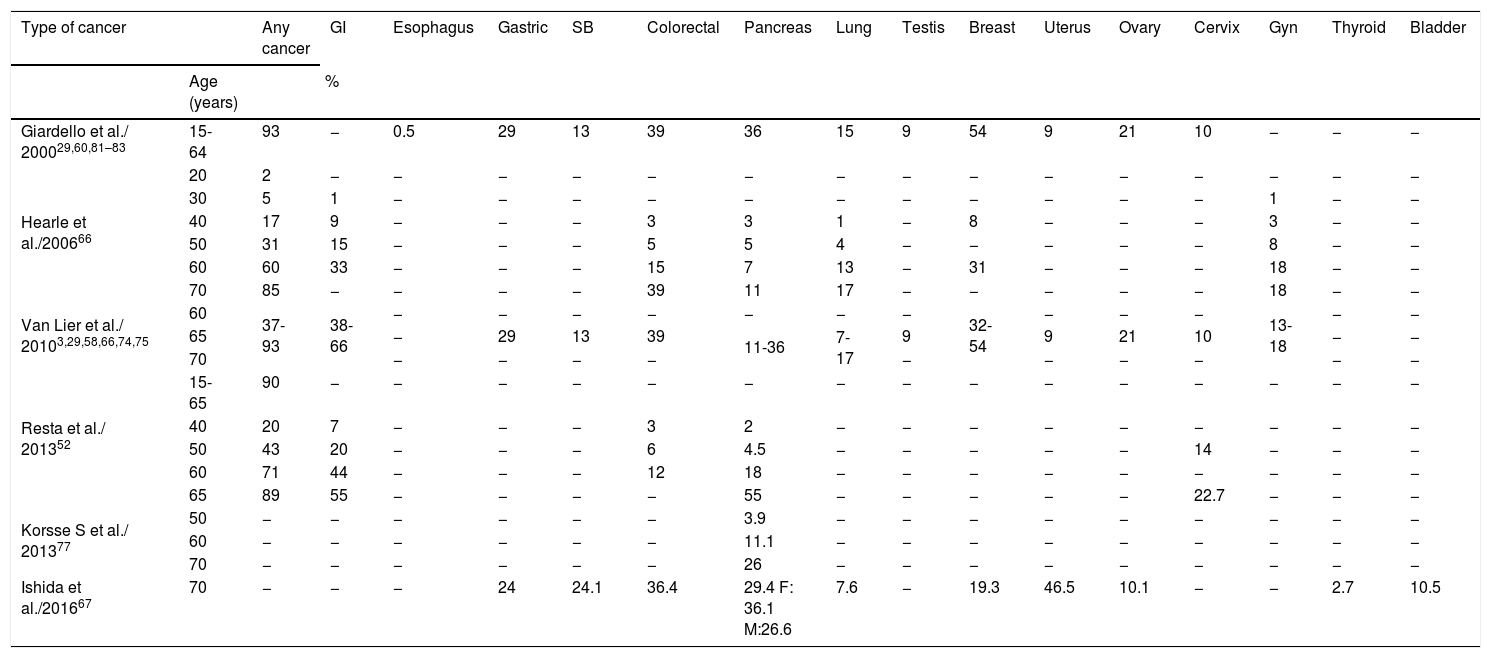
Table 5 show the ARs by patient age reported in different studies. It should be emphasized that in patients 60 to 70 years of age, the AR for all types of cancer ranged from 60% to 93%, for colorectal cancer from 15% to 39%, gastrointestinal cancer from 33% to 66%, and breast cancer from 19% to 54%. In their case series of 419 patients, Hearle et al. (2006) reported that the risk for breast cancer in women increased importantly from 8% to 31% at the ages of 40 and 60 years, respectively.3,29,52,58,66,67,73–75 The considerable rise in the risk for breast cancer can be appreciated, compared with the 12% risk in the Spanish general population.76 In our case series, the percentage of patients that presented with pancreatic cancer was higher than the 5% reported by Korsse et al. (2013) on a cohort of 144 Dutch patients. However, the mean age of 55 years at diagnosis was very similar to our results.77Tables 4 and 5 show the elevated ARs and RRs by age reported by various authors,3,29,52,66,67 especially in patients above 50 years of age. It should be pointed out that Korsse et al. separately classified and analyzed pancreatic cancer, ampullary cancer, and distal bile duct cancer, whereas the authors of the other studies did not specify the anatomic locations.
Accumulated risk for presenting with cancer in patients with PJS.
| Type of cancer | Any cancer | GI | Esophagus | Gastric | SB | Colorectal | Pancreas | Lung | Testis | Breast | Uterus | Ovary | Cervix | Gyn | Thyroid | Bladder | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | % | |||||||||||||||||
| Giardello et al./ 200029,60,81–83 | 15-64 | 93 | − | 0.5 | 29 | 13 | 39 | 36 | 15 | 9 | 54 | 9 | 21 | 10 | − | − | − | |
| Hearle et al./200666 | 20 | 2 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| 30 | 5 | 1 | − | − | − | − | − | − | − | − | − | − | − | 1 | − | − | ||
| 40 | 17 | 9 | − | − | − | 3 | 3 | 1 | − | 8 | − | − | − | 3 | − | − | ||
| 50 | 31 | 15 | − | − | − | 5 | 5 | 4 | − | − | − | − | − | 8 | − | − | ||
| 60 | 60 | 33 | − | − | − | 15 | 7 | 13 | − | 31 | − | − | − | 18 | − | − | ||
| 70 | 85 | − | − | − | − | 39 | 11 | 17 | − | − | − | − | − | 18 | − | − | ||
| Van Lier et al./ 20103,29,58,66,74,75 | 60 | 37-93 | 38-66 | − | − | − | − | − | − | − | 32-54 | − | − | − | 13-18 | − | − | |
| 65 | − | 29 | 13 | 39 | 11-36 | 7-17 | 9 | 9 | 21 | 10 | − | − | ||||||
| 70 | − | − | − | − | − | − | − | − | − | − | ||||||||
| Resta et al./ 201352 | 15-65 | 90 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| 40 | 20 | 7 | − | − | − | 3 | 2 | − | − | − | − | − | − | − | − | − | ||
| 50 | 43 | 20 | − | − | − | 6 | 4.5 | − | − | − | − | − | 14 | − | − | − | ||
| 60 | 71 | 44 | − | − | − | 12 | 18 | − | − | − | − | − | − | − | − | − | ||
| 65 | 89 | 55 | − | − | − | − | 55 | − | − | − | − | − | 22.7 | − | − | − | ||
| Korsse S et al./ 201377 | 50 | − | − | − | − | − | − | 3.9 | − | − | − | − | − | − | − | − | − | |
| 60 | − | − | − | − | − | − | 11.1 | − | − | − | − | − | − | − | − | − | ||
| 70 | − | − | − | − | − | − | 26 | − | − | − | − | − | − | − | − | − | ||
| Ishida et al./201667 | 70 | − | − | − | 24 | 24.1 | 36.4 | 29.4 F: 36.1 M:26.6 | 7.6 | − | 19.3 | 46.5 | 10.1 | − | − | 2.7 | 10.5 | |
F: female, GI: gastrointestinal (colorectal, small bowel, gastric, esophagus, pancreas); Gyn: gynecologic; M: male; SB: small bowel.
It is striking that the ARs for any type of cancer described in 40-year-old patients with PJS ranged from 17% to 20%, when compared with the figures in the 2015 report of the Spanish Network of Cancer Registries. In that report, of all the risks the general population faces for presenting with a certain type of cancer according to location from 0 to 49 years of age, none surpasses the 1.86% that corresponds to the risk for breast cancer in women, signifying that our patients at 40 years of age importantly surpassed the risks for the Spanish general population.78 In addition, in all the case series, the mean ages of the patients at the time of cancer diagnosis range from 36 to 45 years, coinciding with the results of our series and underlining the fact that our case with the earliest age at diagnosis was 30 years.3,13,22,29,39,68 All the above leads us to think that perhaps the presentation of cancer and the ARs for presenting with cancer are higher in young adults, and even higher in the age range of 35 to 50 years. Therefore, the continuous performance of hereditary cancer screening protocols in those patients is extremely important, especially before they reach that age range.
Studies that provide a clinical description related to the presentation of cancer have been carried out, and their results coincide with ours. The percentage of patients in our study that presented with cancer was high, and similar to that recently described by Jelsig et al.20 in their Danish study. They found that 42% of the cases presented with cancer at various anatomic sites. Other cases series reported that 28% to 67% of all the cases presented with cancer.39,50,68 Interestingly, in the study by Tan et al., none of the patients presented with cancer,26 which could possibly be explained by the small number of cases.
Genetic counseling on PJS is extremely beneficial for patients that present with the pathology. To function adequately, it should be carried out by a multidisciplinary team working in conjunction, involving other medical specialties, the clinical laboratory, and the areas of nursing, social work, and psychology. It should also be carried out in accordance with the recommendations and implementation of follow-up and surveillance protocols. Multiple surveillance guidelines have been proposed for cancer screening, the surveillance of polyps, and the integrated management of PJS, and numerous international protocols and recommendations have been described. However, due to the variability of results and the different risk estimates, a consensus has yet to be reached. The Hereditary Cancer in the Valencian Community Guidelines were published in 2008 and updated in 2017.79 Tan et al. reported that only 2/7 cases (28%) received genetic counseling.26 In contrast, in our study, the majority of patients have been seen at an HCGCU, underlining the fact that the Valencian Community is the first autonomous community to regulate the service of an HCGCU.80
ConclusionsThe ICD-9 code 759.6 encompasses many pathologies that are not related to one another, making it nonspecific for the search and register of patients with PJS. The ICD-10 classification may be more specific, given that it includes 3 particular RDs. Nevertheless, those diseases are not related to each other, making another type of classification necessary, one in which there is a code for each disease. It is important to establish standard and unified classifications and codes for RDs, as well as to increase the awareness of how essential their adequate use and implementation are.
The mean age of patients at the time of diagnosis of PJS in the cases from the province of Valencia is within the pediatric population. Therefore, it is necessary to know the characteristics and clinical presentation of PJS in that population.
There is a wide range in the percentage of cases that present with an STK11/LKB1 gene mutation and the percentage of sporadic cases, supporting the genetic heterogeneity of the disease.
Patients with PJS can present with hyperplastic and adenomatous polyps and the finding of an adenomatous polyp can be a red flag.
In the cases of our study, polyps were more frequently located in the SB, especially the duodenum.
PJS is associated with a high risk for presenting with cancer at early ages, as well as with a high complication rate. Intestinal invaginations are a very frequent complication. Surveillance protocols should be reviewed and evaluated to increase the life expectancy of those patients and reduce the presentation of complications.
Screening tests for pancreatic cancer should be included in all the surveillance protocols for PJS.
The implementation and functioning of HCGCUs is beneficial, and they have been well accepted in the management and control of patients with PJS in the province of Valencia in recent years. It would be advantageous for each country to at least unify its protocols based on its population and epidemiologic characteristics, together with the laboratory techniques, diagnostic tests, and treatments utilized. More clinical and epidemiologic studies are needed to determine specific clinical characteristics, risks for associated complications, and therapeutic diagnostic measures in patients with PJS in Spain, as well as studies that evaluate and measure the impact of genetic counseling and surveillance protocols in the Valencian Community on morbidity and mortality.
Financial disclosureNo specific grants were received from public sector agencies, the business sector, or non-profit organizations in relation to this study.
Conflict of interestThe authors declare that there is no conflict of interest.
The authors wish to thank all the patients and their families for accepting to participate in the present study. They also wish to thank all the persons that collaborated from the Administrative Departments of Clinical Documentation, Ethics Committees, Hereditary Cancer Genetic Counseling Units, and Departments of Molecular Biology of all the participating Hospitals and the Hospital General Universitario de Elche, as well as those that collaborated from the Servicio de Estudios Epidemiológicos y Estadísticas Sanitarias de Enfermedades Raras, Servicio de Promoción de Salud y Prevención en el Entorno del Programa de Cáncer Hereditario de la Subdirección General de Epidemiología, Vigilancia de la Salud y Sanidad Ambiental de la Dirección General de Salud Pública de la Comunidad Valenciana. The authors also wish to thank Manuel Posada de la Paz.
Please cite this article as: Rodríguez Lagos FA, Sorlí Guerola JV, Romero Martínez IM, Codoñer Franch P. Registro y seguimiento clínico de pacientes consíndrome de Peutz Jeghers en Valencia. Revista de Gastroenterología de México. 2020;85:123–139.

