
Congenital agastria is an extremely rare condition1 that is frequently associated with other gastrointestinal and extra-gastrointestinal tract malformations.2–4 We present herein the case of a female patient with congenital agastria, with no other associated malformation.
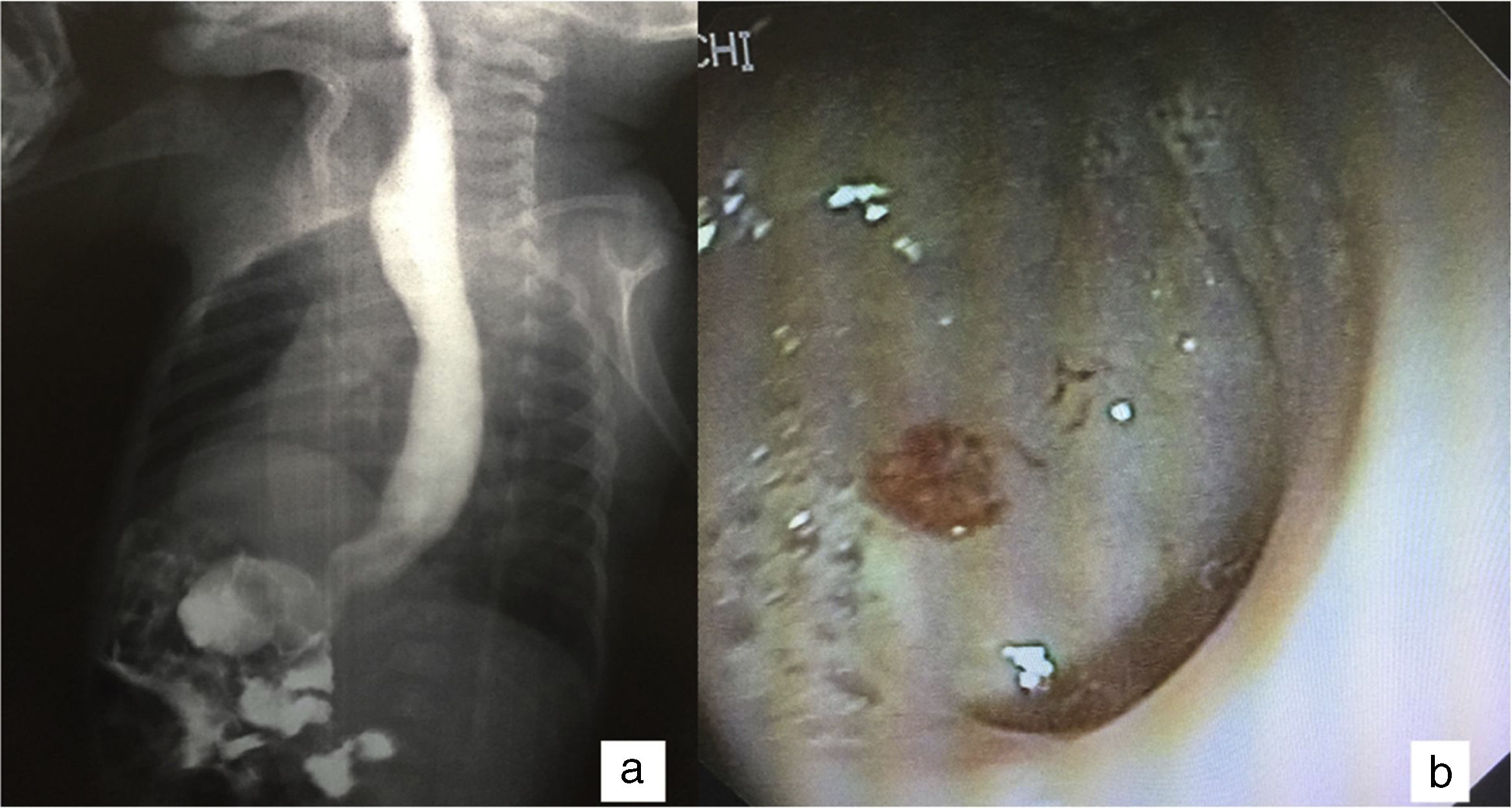
A female infant of 2 months of age was the product of a second pregnancy of 34-week gestation. She was delivered by cesarean section, weighed 2,000g (-3.1 SD, p0) and measured 43cm in height (-3.3 SD, p0), corrected for gestational age. The infant had a history of necrotizing enterocolitis at one month of life and was referred to the Gastroenterology and Nutrition Service of the Instituto Nacional de Pediatría for vomiting and oral diet intolerance. She was managed with prokinetics and exclusion of cow-milk protein, with no improvement. Upon admission, physical examination showed no alterations, with weight of 2.490kg (-5.37 SD, p0) and height of 44cm (-6.42 SD, p0). Contrast-enhanced esophagogastroduodenoscopy revealed dilation of the body of the esophagus and a tubular gastric chamber (fig. 1a). Abdominal ultrasound showed esophageal dilation (transverse diameter: 15mm) and reduced gastric capacity (10ml). Congenital microgastria was suspected. Dilation of the esophageal body was observed in the esophagogastroduodenoscopy, with an absence of epithelial change in the esophagogastric junction and the folds of the gastric corpus. An atrophic and pale mucosal remnant was also observed that was first interpreted as the antral region (fig. 1b). Biopsies of the site were taken that reported superficial esophageal epithelium with hydropic degeneration, supporting the diagnosis of congenital agastria. Echocardiogram, transfontenellar, abdominal, and kidney ultrasound studies, kidney function tests, and x-rays of the spine and extremities were performed to rule out the coexistence of other congenital malformations. A diet based on formula with initial continuous infusion was begun and was adequately tolerated. Once the patient achieved a certain weight recovery, she underwent the creation of a Hunt-Lawrence pouch at 3 months of age. During the intermediate postoperative period, the patient presented with abdominal sepsis. K. pneumoniae BLEE and E. faecalis were isolated and the infant was given broad-spectrum antibiotic therapy. At present, she is 6 months old and is adequately tolerating oral diet and gaining weight in accordance with her corrected gestational age: weight: 5.83 (-0.78 SD, p22), height: 62.2 (0.05 SD, p52).
 EGD study showing the dilation of the body of the esophagus and a tubular gastric remnant; b) endoscopic image of the apparent gastric remnant.")
Congenital agastria is the result of an alteration in the embryogenesis of the stomach. The process begins at the 5th week of fetal life, with the appearance of the gastric primordium (located at the distal part of the anterior intestine), which will later give rise to the stomach. Depending on the time at which that process is interrupted, either complete absence of the stomach or the formation of a small, tubular gastric remnant with minimal functional capacity (microgastria) will be produced.1 The clinical data that those patients commonly present with are postprandial vomiting, gastroesophageal reflux, aspiration pneumonia, and malnutrition. Symptoms vary depending on the phase at which the development of the stomach was detained.2
As previously mentioned, those disorders often are accompanied by other gastrointestinal (esophageal atresia, intestinal malrotation, asplenia, imperforate anus), cardiac, renal, and skeletal abnormalities. Therefore, the initial approach must include the identification of those anomalies.2–4
Diagnosis is suspected through esophagogastroduodenoscopy that commonly reveals a tubular gastric remnant in the midsagittal position, as well as esophageal dilation. However, it is important that biopsies be taken at esophagogastroduodenoscopy, given that the differential diagnosis between agastria and microgastria cannot be made through radiologic or endoscopic images, as was seen with our patient.
Initial treatment is usually medical and consists of ensuring an adequate fluid and electrolyte status and aiding the nutritional recovery of those patients. Nutritional strategies employed for that purpose have been noninvasive (nutrition through continuous infusion utilizing a nasogastric tube) and even surgical (placement of gastrostomy or jejunostomy catheters).2,5,6
The creation of a food reservoir has been shown to be effective for enabling adequate weight gain and improving quality of life in those patients. It can be achieved through the creation of a Hunt-Lawrence pouch, in which an efferent jejunal segment is anastomosed in a laterolateral manner to an efferent jejunal segment, forming a proximal Roux-en-Y pouch.7–9 That technique enables an increase in gastric capacity, reducing the frequency of intakes, improving nutritional ingestion, facilitating bowel transit, and preventing alkaline reflux esophagitis.2 Until 2010, only 13 cases managed through that surgical technique had been reported in the medical literature.1 In cases of isolated congenital agastria, some authors recommend early performance of said procedure, given that it is highly unlikely that the stomach will spontaneously grow.4 Postprandial symptoms during the intermediate postoperative period, such as dumping syndrome, epigastric pain, steatorrhea, fat malabsorption, and bacterial overgrowth, have been reported.4,10 We observed none of those symptoms in our patient. In addition, pernicious anemia prophylaxis with monthly vitamin B12 administration is recommended, due to the lack of intrinsic factor production.
To the best of our knowledge, ours is the first well-documented case of congenital agastria with no other congenital malformations.
Financial disclosureNo financial support was received in relation to this study/article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Cuadros-Mendoza CA, Martínez-Soto MC, Zarate-Mondragón FE, Cervantes-Bustamante R, Ramírez-Mayans JA. Agastria congénita como una malformación aislada. Revista de Gastroenterología de México. 2019;84:258–259.

