
Wilson's disease is characterized by the accumulation of copper in different organs, mainly affecting the liver, brain, and cornea, and is caused by mutations in the ATP7B gene. More than 120 polymorphisms in the ATP7B gene have been reported in the medical literature. The aim of the present study was to identify the conformational changes in the exon 3 region of the ATP7B gene and detect the p.L456V polymorphism in Cuban patients clinically diagnosed with Wilson's disease.
Material and methodsA descriptive study was conducted at the Centro Nacional de Genética Médica and the Instituto Nacional de Gastroenterología within the time frame of 2007-2012 and included 105 patients with a clinical diagnosis of Wilson's disease. DNA extraction was performed through the salting-out method and the fragment of interest was amplified using the polymerase chain reaction technique. The conformational shift changes in the exon 3 region and the presence of the p.L456V polymorphism were identified through the Single-Strand Conformation Polymorphism analysis.
ResultsThe so-called b and c conformational shift changes, corresponding to the p.L456V polymorphism in the heterozygous and homozygous states, respectively, were identified. The allelic frequency of the p.L456V polymorphism in the 105 Cuban patients that had a clinical diagnosis of Wilson's disease was 41% and liver-related symptoms were the most frequent in the patients with that polymorphism.
ConclusionThe p.L456V polymorphism was identified in 64 Cuban patients clinically diagnosed with Wilson's disease, making future molecular study through indirect methods possible.
La enfermedad de Wilson se caracteriza por la acumulación de cobre en diversos órganos: hígado, cerebro y córnea. La causa molecular que la provoca son las mutaciones en el gen atp7b. Se han informado en la literatura más de 120 polimorfismos en el gen atp7b. El objetivo del presente trabajo fue identificar los cambios conformacionales en el exón3 del gen atp7b y detectar el polimorfismo p.L456V en pacientes cubanos con diagnóstico clínico presuntivo de la enfermedad de Wilson.
Material y métodosEn el Centro Nacional de Genética Médica y en el Instituto Nacional de Gastroenterología, durante el período 2007-2012 se realizó un estudio descriptivo que incluyó 105 pacientes con diagnóstico clínico presuntivo de la enfermedad de Wilson. La extracción del ADN fue por la técnica de precipitación salina. Se utilizó la técnica de reacción en cadena de la polimerasa para la amplificación del fragmento de interés, y para identificar los cambios conformacionales y la presencia del polimorfismo p.L456V en el exón 3 del gen atp7b se usó la técnica de polimorfismo conformacional de simple cadena.
ResultadosSe identifican los cambios conformacionales denominados b y c, que correspondieron al polimorfismo p.L456V en estado heterocigótico y homocigótico, respectivamente. La frecuencia alélica del polimorfismo p.L456V en 105 pacientes cubanos diagnosticados clínicamente con la enfermedad de Wilson es del 41%. Las manifestaciones más frecuentes en los pacientes que presentaron este polimorfismo son las hepáticas.
ConclusiónSe identificó el polimorfismo p.L456V en 64 pacientes cubanos con diagnóstico clínico de la enfermedad de Wilson, lo cual posibilitará hacer estudios moleculares por métodos indirectos.
Wilson's disease (WND: MIM# 27790) presents with an autosomal recessive inheritance pattern and is a worldwide health problem. It is classified as a rare genetic disease for which there is treatment, and if untreated, it can cause irreversible damage to the liver and brain that lead to the death of the patient.
The disorder is characterized by an accumulation of copper in different organs: the liver, brain, and cornea. Its clinical diagnosis is complex.1 A distinctive feature of the disease is liver damage, which can manifest through elevated serum transaminase levels to fulminant hepatitis. Patients with WND can suffer alterations at the level of the brain, and can present with psychiatric disorders, such as depression and suicidal tendencies, among others.
The molecular cause of WND are mutations in the ATP7B gene. Said gene has 20 introns and 21 exons that encode for the ATP7B protein, which is the copper transporter in the hepatocyte. More than 120 polymorphisms have been identified that are distributed in the entire ATP7B gene and in the introns. The most polymorphic exons reported are: 2, 8, and 16.2
The p.L456V polymorphism is located in the exon 3 region of the ATP7B gene and is identified in different populations, such as those in China,3 Taiwan,4 Egypt,5 and Venezuela,6 among others. A single nucleotide polymorphism (SNP), it is used in the determination of haplotypes in families with patients that have WND, making their molecular diagnosis possible.6
Adequate screening technology is required to establish the spectrum of polymorphisms in the ATP7B gene. One of the most widely used techniques for that purpose is the single-strand conformation polymorphism (SSCP) analysis. It is based on the relation between the electrophoretic mobility of single-stranded DNA and its three-dimensional structure. A change in the DNA sequence causes a conformational change that in turn causes a change in mobility in the electrophoresis that is detectable by that technique. Thus, conformational changes that are different from the normal variant can be identified in the DNA samples.7
AimsThe aim of the present study was to identify the conformational changes in the exon 3 region and detect the p.L456V polymorphism in Cuban patients with presumptive WND diagnosis.
Materials and methodsA descriptive study was carried out at the Centro Nacional de Genética Médica, within the time frame of 2007-2012, that included 105 patients (43 women and 62 men) with the presumptive diagnosis of WND seen at the Instituto Nacional de Gastroenterología. All the patients consented to participate in the study in accordance with the ethical principles of the Declaration of Helsinki.
The variables analyzed were: allelic frequency of the p.L456V polymorphism, conformational change a for the normal variant, conformational change b for the presence of p.L456V polymorphism in the heterozygous state, and conformational change c for its presence in the homozygous state. The clinical manifestations were classified as hepatic, neurologic, and psychiatric, as well as their combinations.
The clinical manifestations were evaluated by a multidisciplinary team (gastroenterologists, geneticists, neurologists, and biochemists). The established diagnostic criteria for the disease were followed.
The exon 3 region of the ATP7B gene was selected to detect the conformational changes and identification of the p.L456V polymorphism. Blood was drawn from all the patients and DNA extracted through the salting-out method8 from 10ml of peripheral blood with ethylenediaminetetraacetic acid (EDTA) 56mg/ml. The conditions for amplifying exon 3 through the polymerase chain reaction (PCR) were: 100 ng of DNA, 10(F) 5′-AGT CGC CAT GTA AGT GAT AA-3′ and (R) 5′-CTG AGG GAA CAT GAA ACA A-3′, 1mM of dNTPs (Boehringer), 10X PCR buffer, 15mM of MgCl2, 0.3 U of Taq polymerase (Invitrogen), in a volume of 25μl.
SSCP electrophoresis was then performed, mixing 3.5μl with a stop solution of blue bromophenol (0.05% BFA, 10mM NaOH, 95% formamide, 20mM EDTA), and 1μl of the amplified product in a final volume of 7μl, and applied in a gel of commercial acrylamide (GeneGel Excel 12,5/24 Kit). DNA was visualized through the silver staining method, following the instructions of the commercial PlusOne DNA Silver Staining kit (Amersham Biosciences, 2007).
The controls utilized (DNA samples from patients that were heterozygous and homozygous for the p.L456V polymorphism) were donated by Georgios Loudianos, a researcher at the Ospedale Regionale per le Microcitemie laboratory (Cagliari, Italy).
The frequency comparisons were analyzed using the chi-square test.
The present study was approved by the scientific council and the ethics committee of the Centro Nacional de Genética Médica and the Instituto Nacional de Gastroenterología.
ResultsThe mean age of the 105 patients that presented with the p.L456V polymorphism was 31.6 ± 14.1 and the range was 11 years (minimum) and 58 years (maximum).
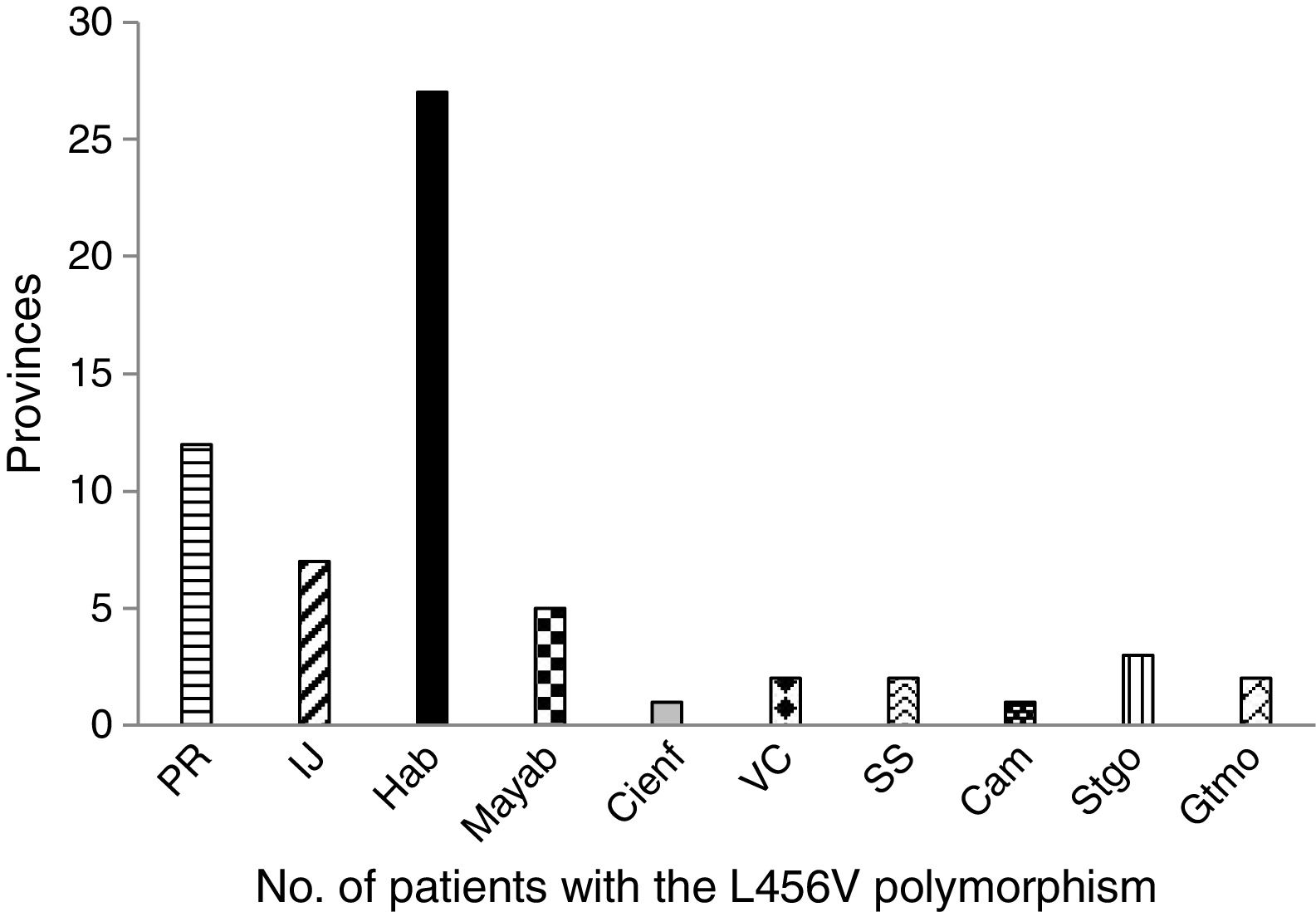
The study patients with the p.L456V polymorphism came from 10 provinces and the special municipality of Isla de la Juventud. The most widely represented province was La Habana, followed by Pinar del Río (fig. 1).

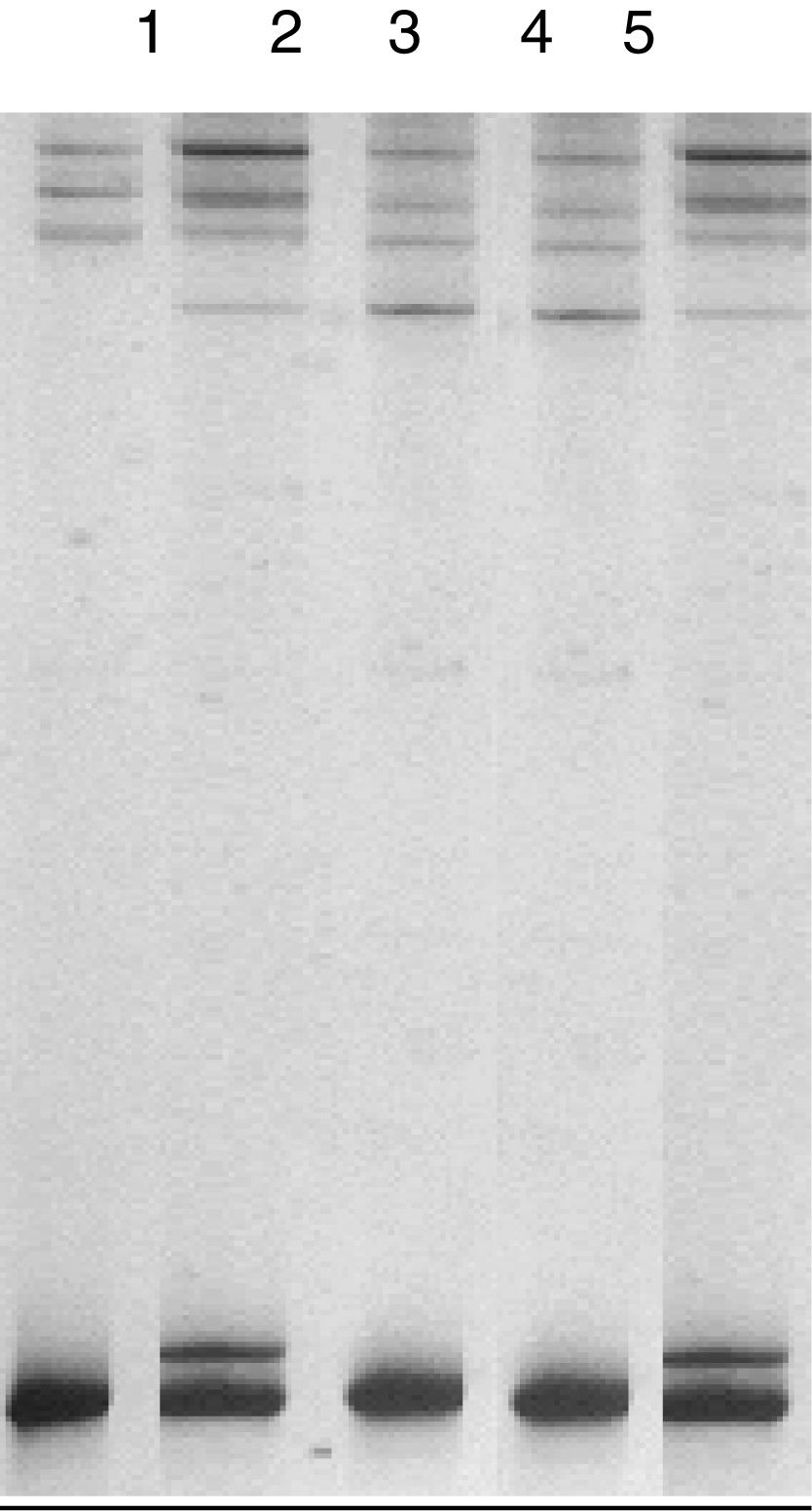
The so-called b and c conformational changes were identified through the SSCP technique (fig. 2) and the so-called a conformational change corresponded to the normal variant. The b and c conformational changes corresponded to the presence of the heterozygous state and the homozygous state of the p.L456V polymorphism, respectively.
 of exon 3 of the ATP7B gene in two positive controls for the p.L456V polymorphism visualized in an acrylamide gel at 12.5%. The ladders correspond to 1: conformational change a, normal variant, negative control; 2: conformational change b, heterozygous control for the p.L456V polymorphism (C-1); 3: conformational change c, homozygous control for the p.L456V polymorphism (C-2); 4: conformational change c, homozygous for the p.L456V polymorphism; 5: conformational change b, heterozygous for the p.L456V polymorphism.")
Single-stand conformation polymorphism (SSCP) of exon 3 of the ATP7B gene in two positive controls for the p.L456V polymorphism visualized in an acrylamide gel at 12.5%. The ladders correspond to 1: conformational change a, normal variant, negative control; 2: conformational change b, heterozygous control for the p.L456V polymorphism (C-1); 3: conformational change c, homozygous control for the p.L456V polymorphism (C-2); 4: conformational change c, homozygous for the p.L456V polymorphism; 5: conformational change b, heterozygous for the p.L456V polymorphism.
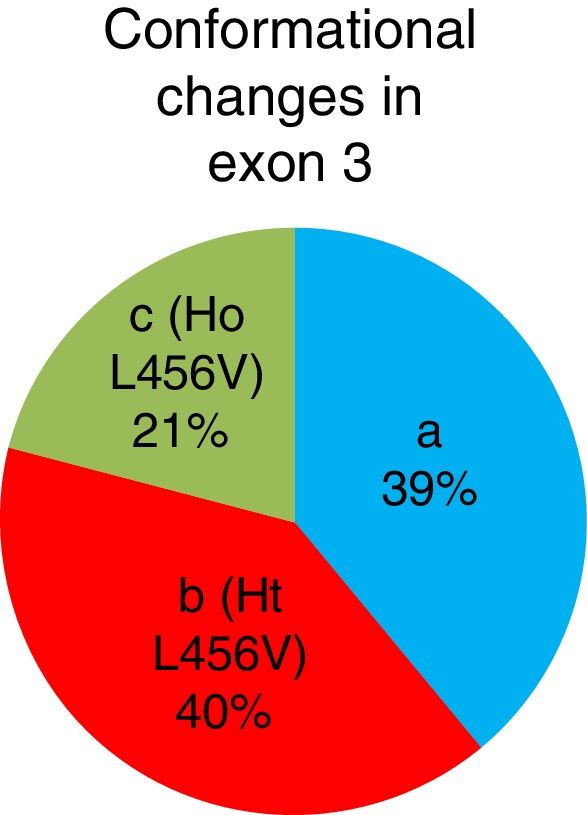
The b conformational change was detected in 42 patients (40%), 22 patients (21%) had the c conformational change, and the a conformational change was detected in 41 patients (39%). A total of 64 patients presented with the p.L456V polymorphism (fig. 3).

Of the 105 Cuban patients with the presumptive clinical diagnosis of Wilson's disease, 42 were determined through the SSCP technique as heterozygous for the p.L456V polymorphism and 22 were homozygous. The allelic frequency of the p.L456V polymorphism of the Cuban patients studied in the present analysis was 41%.
The p.L456V polymorphism is the consequence of the change of guanine for cytosine, causing the amino acid, leucine, to be changed for valine at position 456 of the ATP7B protein. That change does not affect the function of ATP7B, the copper-transporting protein, in humans, and it has been detected in different populations with a frequency above 1%.3–5
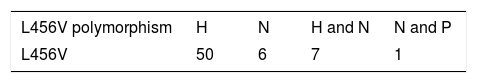
The most frequent clinical manifestations in our study patients that presented with the p.L456V polymorphism were hepatic symptoms (78%), neurologic symptoms (9%), and mixed symptoms (neurologic and psychiatric) (2%) (Table 1).
Main clinical manifestations in Cuban patients presenting with the p.L456V polymorphism.
| L456V polymorphism | H | N | H and N | N and P |
|---|---|---|---|---|
| L456V | 50 | 6 | 7 | 1 |
H: hepatic manifestations; N: neurologic manifestations, H and N: hepatic and neurologic manifestations; N and P: neurologic and psychiatric manifestations.
The presence of Kayser-Fleischer rings, a diagnostic criterion for Wilson's disease, was identified in four patients with the p.L456V polymorphism, representing 6.3% of the study patients. The clinical manifestations of those four patients were: two with hepatic and neurologic symptoms, one with neurologic symptoms, and one with neurologic and psychiatric symptoms.
Discussion and conclusionsThe age of our study patients with the p.L456V polymorphism was similar to that reported in the international literature.
In the distribution by province, La Habana and Pinar de Río had the highest number of patients with the p.L456V polymorphism. The majority of patients with that polymorphism are concentrated in the Western region of the country.
More than 120 polymorphisms in the ATP7B gene have been reported in patients with Wilson's disease.2 In Cuba, detection of the p.L456V polymorphism began in 2007 at the Centro Nacional de Genética Médica. The step before the search for mutations and polymorphisms in the ATP7B gene is the detection of the conformational changes utilizing the SSCP technique. The p.L456V polymorphism was determined by the SSCP comparison of the electrophoretic runs of the positive heterozygous and homozygous controls of the p.L456V polymorphism with the PCR product obtained from the exon 3 region of the ATP7B gene from the samples of the Cuban patients analyzed.
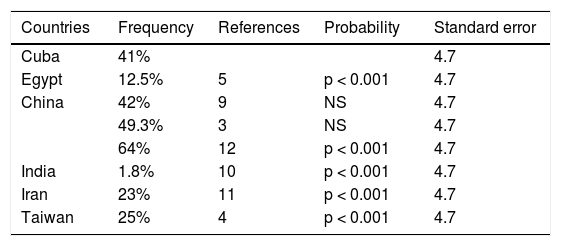
The frequency of the p.L456V polymorphism in two Chinese studies was similar to that of our analysis (there were no statistically significant differences), and as expected, frequency was above 1%. There were statistically significant different when our results were compared with those conducted in Egypt, India, Iran, and Taiwan. A possible explanation for that is the ethnic origin of the Cuban population. As shown in Table 2, our result is the second highest in frequency of those reported in the countries analyzed. Future studies will evaluate the impact of the p.L456V polymorphism in Cuban patients with a molecular diagnosis of WND. It was interesting to observe that the frequency of the p.L456V polymorphism was very low in India (1.8%), where it is as a rare variant.
Frequencies by country of the p.L456V polymorphism in patients with the clinical diagnosis of Wilson's disease.
| Countries | Frequency | References | Probability | Standard error |
|---|---|---|---|---|
| Cuba | 41% | 4.7 | ||
| Egypt | 12.5% | 5 | p < 0.001 | 4.7 |
| China | 42% | 9 | NS | 4.7 |
| 49.3% | 3 | NS | 4.7 | |
| 64% | 12 | p < 0.001 | 4.7 | |
| India | 1.8% | 10 | p < 0.001 | 4.7 |
| Iran | 23% | 11 | p < 0.001 | 4.7 |
| Taiwan | 25% | 4 | p < 0.001 | 4.7 |
Studies have been conducted in several countries that carried out molecular analyses in patients with WDN, but they did not report the presence of the p.L456V polymorphism.9,10 In Turkey, the p.L456V variant was not identified, but the p.V446L polymorphism was found in the exon 3 region (where the p.L456V polymorphism is located).11
Advanced technology is required for successfully achieving the molecular diagnosis of genetic diseases that possess one or more genes with an elevated number of exons. Different molecular studies have been conducted on patients with WND,12,13 but the physician treating the patient with suspicion of WND must always perform a detailed clinical diagnosis, taking the criteria established worldwide into account.14
The p.L456V polymorphism has been identified in different countries, but not its frequencies,6,15 making the comparison of our results impossible. In some cases, haplotypes are not constructed with the use of different SNPs determined in their studies and thus do not take advantage of the opportunity to broaden the molecular diagnosis.
Even when new mutations are determined16–19 and techniques with microarrays are carried out20 in developing countries, it is necessary that genetic diseases are studied through molecular biology and that the search for polymorphisms is included to provide the service of molecular diagnosis through indirect methods in diseases that present with a considerable number of exons and few frequent mutations.
The establishment of a strategy for the molecular diagnosis of WND is being worked on in Cuba. Our work team has experience in identifying polymorphisms in exons 1021 and 1322,23 and in intron 9.24 With the polymorphisms that have already been detected, and now with the p.L456V polymorphism in Cuban patients with the presumptive diagnosis of WND, haplotypes can be determined in the Cuban families with that disease, as well as molecular diagnosis through indirect methods. In addition to the detection of the p.L456V polymorphism, different mutations have been reported in China,25–26 India,27 and Iran28 for establishing the molecular diagnosis of WND, making molecular diagnosis through indirect and direct methods possible, carrying out their strategies for the sake of improving the quality of life of the patients with WND.
In relation to the ethnic origin of the Cuban population, it is important to point out that the islands of the Caribbean, including Cuba, were the first to be inhabited by Mesoamericans and later by Amerindians, which some researchers have suggested came from Venezuela.29,30 It is estimated that the island had more than 100,000 inhabitants when the Spanish arrived and during the first 50 years of the Conquest, the population was reduced to about 5,000 individuals. The Spanish colonizers extinguished the Amerindian population and began to bring aborigines from North American and Mesoamerica to Cuba, as well as slaves from the West coast of Africa and black slaves from East Africa and Sub-Saharan Africa. Thus, the genetic composition of the Cuban population is basically a mixture of Caucasian Spaniards and black Africans.31,32 The Spanish immigration remained constant from the end of the fifteenth century to the first half of the twentieth century. The history of the ethnic miscegenation that took place between those three original groups (Native Americans, Europeans, and Africans) for nearly five centuries has created the genetic structure of the current Cuban population. The average contribution of the genes of European, African, and Native American origin in the individuals studied was 72, 20, and 8%, respectively.32 Ethnicity was observed to play an important role in the distribution of the mutations in the ATP7B gene that are responsible for WND.33 Future study of the mutations and polymorphisms of the ATP7B gene in the American continent and the evaluation of the role of ethnicity in their presence, as well as in the susceptibility to disease, will be useful.
In conclusion, a molecular tool is available for introducing the molecular diagnosis in patients with WND in Cuba. The methodology can be used in other population with a genetic profile similar to that of Cuba.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that the procedures followed were in accordance with the regulations of the relevant clinical research ethics committee and with those of the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors have obtained the written informed consent of the patients or subjects mentioned in the article. The corresponding author is in possession of this document.
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
The authors wish to thank the patients for participating in the present study, Lídice Reyes for performing the DNA extraction, The Public Health Ministry, Dr. Carlos Maragoto for his contributions, Georgios Loudianos for the delivery of the positive controls, and Dr. Carlos Castañeda for his contributions with respect to the clinical diagnosis of the disease.
Please cite this article as: Clark-Feoktistova Y, Ruenes-Domech C, García-Bacallao EF, Roblejo-Balbuena H, Feoktistova L, Clark-Feoktistova I, et al. Polimorfismo p.L456V, presencia en pacientes cubanos con diagnóstico clínico presuntivo de la enfermedad de Wilson. Revista de Gastroenterología de México. 2019;84:143–148.
The present study is the result of the joint work of the Centro Nacional de Genética Médica (a center collaborating with the WHO for the development of genetic approaches to health promotion), the Instituto Nacional de Gastroenterología, and the Universidad de Guantánamo.


www.publicationethics.org.