
Alcoholic liver disease (ALD) is a clinical-pathologic entity caused by the chronic excessive consumption of alcohol. The disease includes a broad spectrum of anomalies at the cellular and tissual level that can cause acute-on-chronic (alcoholic hepatitis) or chronic (fibrosis, cirrhosis, hepatocellular cancer) injury, having a great impact on morbidity and mortality worldwide. Alcohol is metabolized mainly in the liver. During alcohol metabolism, toxic metabolites, such as acetaldehyde and oxygen reactive species, are produced. At the intestinal level, alcohol consumption can cause dysbiosis and alter intestinal permeability, promoting the translocation of bacterial products and causing the production of inflammatory cytokines in the liver, perpetuating local inflammation during the progression of ALD. Different study groups have reported systemic inflammatory response disturbances, but reports containing a compendium of the cytokines and cells involved in the pathophysiology of the disease, from the early stages, are difficult to find. In the present review article, the role of the inflammatory mediators involved in ALD progression are described, from risky patterns of alcohol consumption to advanced stages of the disease, with the aim of understanding the involvement of immune dysregulation in the pathophysiology of ALD.
La enfermedad hepática alcohólica (EHA) es una entidad clínico-patológica, ocasionada por el consumo excesivo y crónico de alcohol. La enfermedad incluye un amplio espectro de anomalías a nivel celular y tisular que pueden causar daño agudo sobre crónico (hepatitis alcohólica) o crónico (fibrosis, cirrosis, cáncer hepatocelular), teniendo un gran impacto en la morbilidad y mortalidad a nivel mundial. El alcohol es metabolizado principalmente en el hígado. Durante el metabolismo del alcohol son generados metabolitos tóxicos como el acetaldehído y las especies reactivas de oxígeno. A nivel intestinal, el consumo de alcohol puede producir disbiosis y alteración de la permeabilidad intestinal, promoviendo la translocación de productos bacterianos, y provocando la producción de citocinas proinflamatorias en el hígado, lo cual perpetua la inflamación local durante la evolución de la EHA. Diferentes grupos de estudio han reportado alteraciones de la respuesta inflamatoria a nivel sistémico, sin embargo, es difícil encontrar reportes que contengan un compendio de las células y citocinas involucradas en la fisiopatología de la enfermedad desde sus etapas tempranas. En este trabajo de revisión se describe el papel de los mediadores inflamatorios involucrados en la progresión de la EHA, desde los patrones de consumo de riesgo hasta etapas avanzadas de la enfermedad, con la finalidad de comprender la implicación de la desregulación inmunológica en la fisiopatología de esta enfermedad.
Alcoholic liver disease (ALD) is a clinical-pathologic entity caused by excessive alcohol consumption and can include a spectrum of entities, such as steatosis, steatohepatitis, fibrosis, cirrhosis, hepatocellular carcinoma, and in some cases, alcoholic hepatitis (AH). Progression from one entity to another depends on the continuation of alcohol consumption and the complex interaction of a large number of variables: genetic, biologic, immunologic, psychologic, sociocultural, and environmental. Nevertheless, its pathogeny is still not completely understood.1,2
In Mexico, the results of the 2016-2017 National Survey on Drug, Alcohol, and Tobacco Consumption (ENCODAT, its Spanish acronym) reveal that alcohol consumption has risen in the Mexican population between 18 and 65 years of age, with a marked increase of alcohol dependence in adolescent girls.3 Age at first alcohol use went from 17 to 16 years, reflecting the fact that adolescents are a risk group for developing disorders related to alcohol abuse.3
Alcohol consumption in those age groups is a risk factor for developing addiction, given that it induces alterations involving the peri-frontal region of the brain, the area that is in charge of behavioral regulation and planning.4,5 Regarding genetic influence, ADH1B and ALDH2 have been described as the 2 main genes that participate in alcohol metabolism, and they are related to excessive consumption and alcohol dependence. Information about those genes and their genetic variants is excellently described in specialized reviews on the theme6,7 but they are not yet used as a diagnostic tool for the different patterns of alcohol consumption.
In Mexico, ALD is the most frequent cause of cirrhosis of the liver.8 Up to June 2021, the incidence rate of alcoholic liver cirrhosis was 2 cases per 100,000 inhabitants, situating Mexico in fifth place for mortality due to liver disease9; according to disease projection studies, the mortality rate is expected to be on the rise in the coming years.10
The age group most affected by cirrhosis due to ALD is that of 51 to 70 years, due to the fact the disease has usually reached advanced stages by the time medical attention is sought.11 Because of active alcohol consumption in the Mexican population and the low rate of screening for risky alcohol consumption, and consequently, the development of ALD, the number of cases of liver damage and cirrhosis is predicted to rise in the near future, increasing the cirrhosis mortality rate.8 Various authors have made extensive and detailed epidemiologic descriptions in Mexico and across the globe.12–14 Worldwide, the majority of cases of ALD are diagnosed at advanced stages of the disease, signifying cirrhosis of the liver.11,15 At present, questionnaires enabling a psychiatric assessment16 are utilized to evaluate alcohol consumption but there is no objective and quantitative tool available.
ALD can present with acute and chronic stages, or a combination of the two, and so the local (liver) and systemic (blood circulation) response actively participates, according to the frequency and type of alcohol consumed.1,17 In the present review article, the pathophysiologic effects in ALD and the immunologic alterations during disease progression are emphasized. In addition, the alcohol consumption classification criteria are presented, and even though they are not yet completely defined, they are of vast importance for the early diagnosis of disease resulting from alcohol abuse.
Alcohol consumption patternThe standard drink now has various definitions worldwide, and so determining the quantity and frequency of consumption tends to be difficult. In Mexico, the standard drink is that which contains 13 grams of alcohol.18 Regarding the type of alcoholic beverage consumed, the results of the ENCODAT revealed that fermented drinks, specifically beer, hold first place in prevalence, at 40.8%, and distilled drinks (brandy, tequila, rum, whisky, cognac, vodka, etc.) hold second place, at 19.1%.3 Alcoholic beverage quality is still a subject of debate, given that standards are not regulated. Lastly, clinical effects and liver injury tend to be evident until advanced stages of ALD.
Moderate alcohol consumption has been associated with protective effects at the cardiovascular level, with antidepressant effects, and with reducing cerebrovascular accidents.19–21 Moderate consumption is defined as the intake of one standard drink per day for women and 2 for men.22 Risky alcohol consumption is considered if the individual has a score of 8 or higher on the alcohol use disorders identification test (AUDIT) questionnaire,23 and ingests 40-60 g (for men) or 20-40 g (for women).24 Alcohol consumption abuse is usually considered when the AUDIT questionnaire score is 8 or higher and the diagnostic and statistical manual of mental disorders (DSM)-IV abuse criteria are met.25 That pattern is characterized by intake higher than 60 g (for men) or higher than 40 g (for women), in addition to binge drinking (consumption > 70 g for men and > 50 g for women), one to 4 times a week.
According to the World Health Organization (WHO), alcoholism is defined as the daily intake of more than 70 g (for men) and 50 g (for women) for at least 5 years, with an AUDIT score above 8, and meeting the DSM-IV dependency criteria.26–28 Today, it is common to find subjects diagnosed as alcoholics if they consume those quantities on the week-ends or on binges.29 Importantly, clinical and biochemical parameters have no obvious alterations in individuals that have risky consumption patterns, alcohol abuse, or alcoholism, and so they rarely seek medical attention.11
Among the current tools for diagnosing problems of alcohol consumption are the AUDIT, CAGE (Cut down, Annoyed, Guilty, Eye-opener), and the DSM. However, at present, there are no biochemical parameters or specific markers that enable the quantitative determination of the effect of harmful alcohol consumption from an early stage, such as that of liver steatosis. Patients are frequently asymptomatic until they develop a severe or advanced stage of disease.1
Our research group has found that individuals that have already developed ALD (cirrhosis or AH) more frequently consume drinks that are distilled, made with cane alcohol and/or 96° alcohol (work in process). The Maddrey score is the most widely used scoring system in AH and is based on bilirubin and prothrombin time values of the patient at hospital admission.30,31 It functions as a guide for treatment and a predictor of disease mortality and severity.30,31 A score > 32 indicates severe disease, with a high mortality rate (30-60%) in the short term, whereas a score < 32 indicates nonsevere disease, with a short-term mortality rate of 10%.30,31 In addition, cirrhosis of the liver due to alcohol is a chronic condition that presents characteristic signs, such as sarcopenia, weight loss, asthenia, adynamia, hepatojugular reflux, telangiectasias, gynecomastia, and palmar erythema, among others.1 Nevertheless, some patients with cirrhosis are diagnosed when they develop liver decompensation, frequently in the context of overlap with AH.1 The Child-Pugh classification has been widely used for evaluating survival in patients with cirrhosis.32 Child-Pugh class A is a score of 5-6 points, class B is 7-9 points, and class C is 10-15 points. Class A has the highest one-year survival (100%) and 2-year survival (85%) and class C has the worst one-year survival (45%) and 2-year survival (35%).32
Pathophysiology of alcoholic liver diseaseAlcohol is primarily metabolized in the stomach and intestine, where toxic metabolites, such as acetaldehyde, an essential component for progression to ALD, are formed.33,34 The alcohol then passes into the blood stream and portal circulation, to arrive at the liver, where the greater part of metabolism takes place, through the hepatocytes. If consumption is excessive, the microsomal pathway is activated, which can produce reactive oxygen species (ROS).33,35,36
During ALD, steatosis presents in around 90% to 100% of alcohol consumers, and 35% of them progress to steatohepatitis. When excessive alcohol consumption continues, liver fibrosis is developed, which can advance to cirrhosis in around 20% of cases, and 1% to 2% of those patients will develop hepatocellular carcinoma.17 Nevertheless, the disease stages are not separate, but rather can coexist in the same person.17,37 AH can develop in 40% of the subjects with cirrhosis, but can even appear as a result of steatohepatitis due to excessive alcohol consumption.2,17 AH is a severe complication that can occur at any point during the course of the disease and is associated with liver failure, with a short-term mortality rate of up to 40%.38,39 Despite the fact that there are well-defined stages, the complex interaction between the cellular and molecular factors of ALD is not yet fully understood.2,17
The gut-liver axisExcess consumption of alcohol can cause deficient digestion, poor nutrient absorption, vitamin and trace element deficiencies, and in turn, weight loss, mainly due to the first step of alcohol metabolism.40 The toxicity of alcohol comes primarily from the metabolic product of alcohol, acetaldehyde, that can also be produced by several bacteria in the colon, leading to colonic mucosal cell injury.41
At the level of the colon, acetaldehyde can cause disruption in the tight junction proteins, promoting intestinal barrier dysfunction.41 In addition, alcohol consumption can affect intestinal motility, the pH of the intestinal lumen, and bile flow.42
SteatosisAt the hepatic level, the initial alteration in ALD is steatosis, due to lipid uptake deregulation.17,43 Alcohol can induce steatosis through the following disruptions:
- -
Increase in the proportion of NADH/NAD+ in the hepatocytes, interrupting fatty acid β-oxidation at the mitochondrial level.44
- -
Increase in the hepatic expression of the sterol regulatory element-binding protein-1c (SREBP1c), stimulating the expression of lipogenic genes and fatty acid synthesis.45,46 Alteration of the transcription of SREBF1 that encodes SREBP1c and PPARα that encodes the peroxisome proliferator-activated receptor alpha (PPARα) can occur directly due to acetaldehyde, but it can also be activated by bacterial translocation.47
- -
Inhibition of the adenosine monophosphate (AMP)-activated kinase protein, promoting fatty acid synthesis and inhibiting β-oxidation through the deregulation of acetyl-CoA carboxylase, the hepatic isoform of carnitine palmitoyltransferase 1, and SREBP.48
- -
Induction of lipolysis and destruction of adipocytes, resulting in the increase in circulating fatty acids and their later accumulation in the liver.43,49,50
Another well-defined stage is alcoholic steatohepatitis, characterized by the presence of lipids and an obvious inflammatory reaction, with a predominance of neutrophils, ballooning degeneration of hepatocytes, Mallory-Denk body inclusions, and a perivenular fibrosis network.51 It should be pointed out that the inflammatory reactions are induced since the stage of steatosis, but they have a marked increase during alcoholic steatohepatitis.52 Immunologic activity produces endoplasmic reticulum stress, activating interferon regulatory factor 3 (IRF3). However, the IRF3 molecule is phosphorylated when exposed to alcohol, activating hepatocyte apoptosis.41 Kupffer cells are activated by lipopolysaccharide (LPS) translocation, promoting neutrophil recruitment, because the Kupffer cells secrete proinflammatory cytokines, such as TNF-α, as well as the chemokine, CXCL-8.53
Factors that contribute to the development of fibrosis and cirrhosisFibrosis is the stage characterized by an uncontrolled accumulation of extracellular matrix at the pericellular and perisinusoidal level, as a response to persistent inflammation and injury.1 The large quantity of cytokines and growth factors secreted by the Kupffer cells promotes hepatic stellar cell (HSC) activation, which is the key event in liver fibrogenesis. Acetaldehyde itself is capable of directly inducing HSC activation.54,55 Excessive alcohol consumption intervenes in HSC regulation by suppressing the cytotoxic activity of the natural killer (NK) cells toward activated HSCs and their secretion of IFN-γ that induces activated HSC apoptosis, favoring the excessive production of extracellular matrix by the HSCs.56
Nevertheless, liver fibrosis is multifactorial. Among the factors involved in its development are the ROS generated by alcohol metabolism that produce lipid peroxidation. The metabolites malondialdehyde and 4-hydroxynonenal are products of lipid peroxidation and they induce the formation of neoantigens, altering the immune response.57 Acetaldehyde enters the HSCs in a paracrine manner and induces transforming growth factor beta-1 (TGF-β1) expression, consequently stimulating type 1 collagen production.57 Acetaldehyde phosphorylates the SMAD 3 protein, activating the SMAD 3-4 complex, which is how TGF-β and acetaldehyde form a positive feedback loop of collagen genes, especially the type 1 collagen α2 gene.58
Hepatocyte apoptosis has been shown to play a role in the appearance of fibrosis. The apoptotic bodies release lipid signals for their processing by Kupffer cells and HSCs, promoting the expression of profibrogenic genes, such as TGF-β1, that trigger HSC activation.58 The above-described mechanisms can diminish if the patient stops consuming alcoholic beverages, but if the inflammatory process continues, it progresses to a chronic stage with sustained fibrogenesis, resulting in the substitution of the liver parenchyma by scar tissue. That compromises the architecture and vascular network of the liver, developing cirrhosis that is characterized by the formation of regenerative nodules of the hepatic parenchyma that are surrounded by fibrotic septa.59,60 Its development is a compensated phase, in which part of the liver remains intact and functionally compensates the injured regions. Posteriorly, it advances to a decompensated phase, in which the scar tissue completely envelops the organ.61,62 The latter phase is characterized by the development of portal hypertension, with or without the presence of liver failure.63
Anti-inflammatory use in alcoholic liver diseaseAt present, there is no treatment involving anti-inflammatory molecules for alcohol-induced liver disease.64 AH is the most widely studied entity due to its high mortality in a short period of time (28 days). For more than 50 years, corticosteroids have had a central role, but their benefit in treatment has become controversial.65 The largest randomized clinical trial on AH, “Steroids or Pentoxifylline for Alcoholic Hepatitis” (STOPAH), showed no statistically significant benefit in 28-day survival in patients that received corticosteroids, compared with placebo (p = 0.06). The current guidelines on AH management recommend the oral administration of 40 mg of prednisone daily to reduce 28-day mortality in patients with severe AH.66 Given the abovementioned, there is an unavoidable need to improve the treatment of AH and ALD. In that sense, IMM-124E is a purified, hyperimmune bovine colostrum formulation, which contains the IgG antibody against LPS, that was developed to reduce endotoxemia. The results of the randomized clinical trial, NCT01968382, revealed no efficacy as an adjuvant therapy to corticoids in patients with severe AH.65 In phase II and III clinical trials (NCT01809132), treatment with the IL-1 antagonist receptor, anakinra, in combination with pentoxifylline and zinc sulfate, was compared with treatment with methylprednisolone,67 revealing no improvement in survival with the therapeutic combination evaluated.67,68
The use of TNF-α antagonists, such as infliximab and etanercept, has been associated with an increase in mortality and incidence of infections,69,70 and so the present day guidelines do not recommend them.66 Recently, a phase II clinical trial utilizing F652 (a recombinant protein) showed a decrease in the MELD score and inflammation markers, as well as an increase in liver regeneration markers in patients with AH.71
Among the therapies that have a potential benefit, with clinical efficacy, are those with antioxidants, such as N-acetylcysteine (NAC) and metadoxine, albeit they have not been fully tested.65,66,72–74 In a randomized clinical trial on patients with severe AH, the infusion of NAC, as an adjuvant therapy to corticosteroids, improved survival at one month, but there was no long-term benefit, mainly due to the increase in infection rates and the development of hepatorenal syndrome.73 On the other hand, metadoxine, as an adjuvant therapy to corticosteroids or to pentoxifylline, was associated with survival improvement at 3 and 6 months in patients with severe AH.74
Lastly, liver transplantation is the indicated procedure for advanced liver injury, but a high percentage of patients die before getting on the waiting list.10,75,76
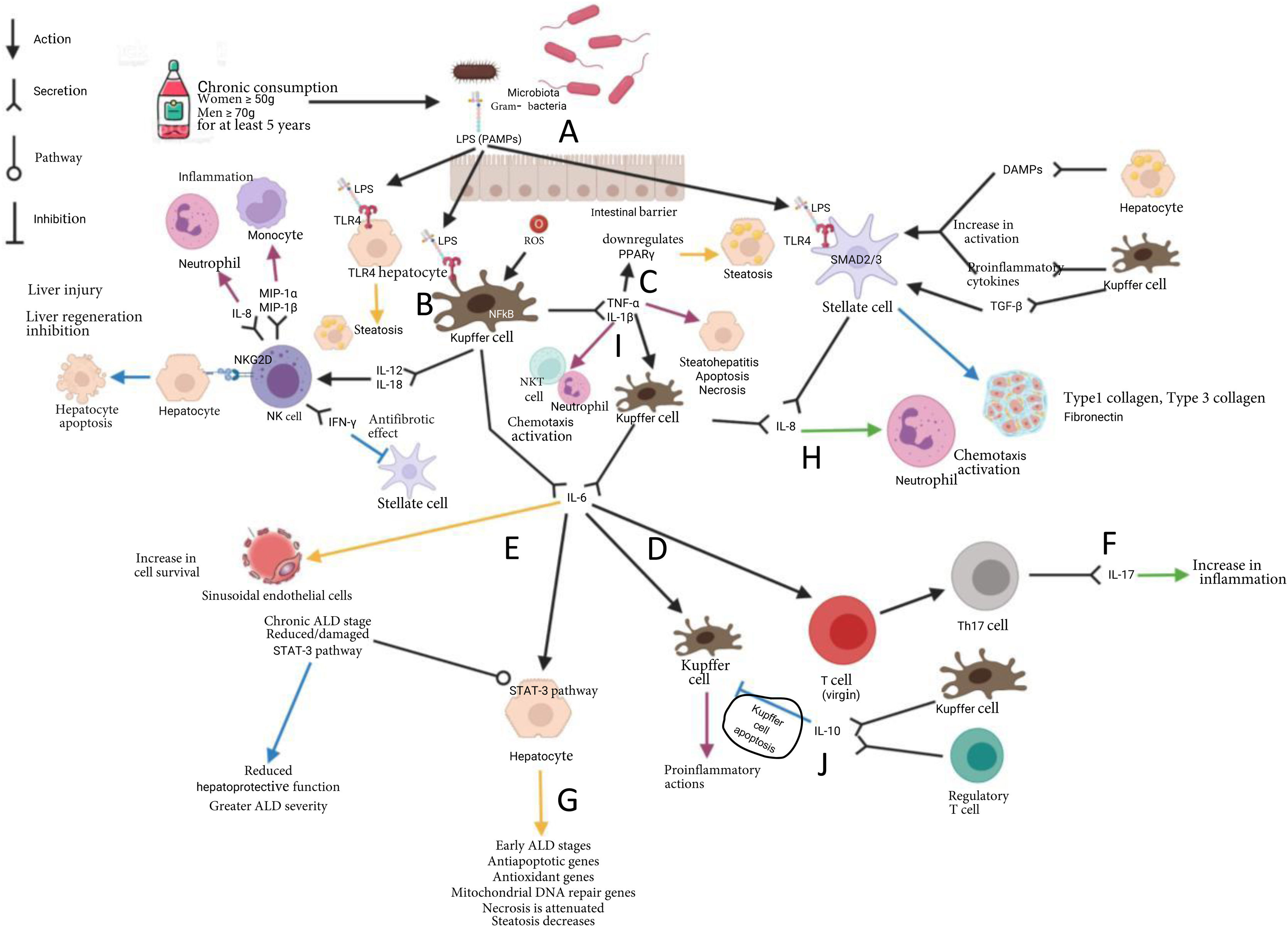
Inflammation during alcoholic liver disease progressionThe chronic consumption of alcohol leads to intestinal dysbiosis, causing a change of dominance into Gram (-) and opportunistic agents. Likewise, it causes a decrease of the microorganisms that characterize the intestinal mucosa of healthy subjects.40 The duodenal mucosa of subjects with chronic alcohol consumption presents with a decrease in inflammatory cells, such as macrophages and T lymphocytes. Thus, there is an attenuated inflammatory response, which could be a permissive factor for bacterial translocation.40 At the same time, there is a disruption of the tight junction proteins in the intestinal barrier, resulting in increased intestinal permeability40 (Fig. 1A).
 bacteria (A). The chronic ingestion of alcohol gives rise to the disruption of the intestinal barrier, causing an increase in intestinal permeability, enabling the translocation of LPS and its arrival at the liver through the portal circulation. In the liver, LPS binds to TLR4 in the Kupffer cells (B), that together with ROS, stimulate TNF-α and IL-1β synthesis. TNF-α downregulates PPARγ gene expression, favoring the expression of the genes that participate in β-oxidation, contributing to the development of steatosis (C). TNF-α and IL-1β give rise to the production of IL-6 by the Kupffer cells, thus perpetuating the inflammatory state (D). The production of IL-6 by the Kupffer cells plays a dual role, favoring inflammation through the positive feedback loop of the Kupffer cells (E) and activation of the Th17 response (F). On the other hand, IL-6 favors the activation of STAT-3 in the early stages of ALD, resulting in a hepatoprotective effect (G). IL-8 causes chemotaxis and neutrophil activation, leading to inflammation and tissue damage (H). Stellate cell stimulation by PAMPs, DAMPs, and Kupffer cells favors their activation and the later production of IL-8 and components of the extracellular matrix, giving rise to the development of fibrosis (I). IL-10 negatively regulates the inflammatory responses through promoting the apoptosis of macrophages with an inflammatory profile (J). Kupffer cells, activated through the binding of ligands to their TLR4 receptor, induce the recruitment and activation of NK cells, through IL-12 and IL-18 production; IL-12 and IL-18 are cytokines that strikingly induce the production of IFN-y by NK cells, which has an antifibrotic effect upon inducing apoptosis and arresting the HSC cell cycle, as well as increasing NK cell cytotoxicity (K). Yellow arrow: steatosis; Purple arrow: steatohepatitis; Blue arrow: fibrosis; Green arrow: alcoholic hepatitis. ALD: alcoholic liver disease; DAMPs: danger associated molecular patterns; DNA: deoxyribonucleic acid; IFN-γ: interferon gamma; IL: interleukin; LPS: lipopolysaccharide; MIP-1α: macrophage inflammatory protein 1 alpha; MIP-1β: macrophage inflammatory protein 1 beta; NF-kB: nuclear factor kappa B; NK: natural killer; NKG2D: natural killer group 2D receptor; NKT: natural killer T; PAMPs; pathogen-associated molecular patterns; PPARα: peroxisome proliferator-activated receptor-alpha; ROS: reactive oxygen species; SMAD: Smad family; TGF-β: transforming growth factor beta; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor alpha; STAT3: signal transducer and activator of transcription 3.")
Chronic alcohol consumption causes intestinal dysbiosis, favoring the proliferation of Gram (-) bacteria (A). The chronic ingestion of alcohol gives rise to the disruption of the intestinal barrier, causing an increase in intestinal permeability, enabling the translocation of LPS and its arrival at the liver through the portal circulation. In the liver, LPS binds to TLR4 in the Kupffer cells (B), that together with ROS, stimulate TNF-α and IL-1β synthesis. TNF-α downregulates PPARγ gene expression, favoring the expression of the genes that participate in β-oxidation, contributing to the development of steatosis (C). TNF-α and IL-1β give rise to the production of IL-6 by the Kupffer cells, thus perpetuating the inflammatory state (D). The production of IL-6 by the Kupffer cells plays a dual role, favoring inflammation through the positive feedback loop of the Kupffer cells (E) and activation of the Th17 response (F). On the other hand, IL-6 favors the activation of STAT-3 in the early stages of ALD, resulting in a hepatoprotective effect (G). IL-8 causes chemotaxis and neutrophil activation, leading to inflammation and tissue damage (H). Stellate cell stimulation by PAMPs, DAMPs, and Kupffer cells favors their activation and the later production of IL-8 and components of the extracellular matrix, giving rise to the development of fibrosis (I). IL-10 negatively regulates the inflammatory responses through promoting the apoptosis of macrophages with an inflammatory profile (J). Kupffer cells, activated through the binding of ligands to their TLR4 receptor, induce the recruitment and activation of NK cells, through IL-12 and IL-18 production; IL-12 and IL-18 are cytokines that strikingly induce the production of IFN-y by NK cells, which has an antifibrotic effect upon inducing apoptosis and arresting the HSC cell cycle, as well as increasing NK cell cytotoxicity (K).
Yellow arrow: steatosis; Purple arrow: steatohepatitis; Blue arrow: fibrosis; Green arrow: alcoholic hepatitis.
ALD: alcoholic liver disease; DAMPs: danger associated molecular patterns; DNA: deoxyribonucleic acid; IFN-γ: interferon gamma; IL: interleukin; LPS: lipopolysaccharide; MIP-1α: macrophage inflammatory protein 1 alpha; MIP-1β: macrophage inflammatory protein 1 beta; NF-kB: nuclear factor kappa B; NK: natural killer; NKG2D: natural killer group 2D receptor; NKT: natural killer T; PAMPs; pathogen-associated molecular patterns; PPARα: peroxisome proliferator-activated receptor-alpha; ROS: reactive oxygen species; SMAD: Smad family; TGF-β: transforming growth factor beta; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor alpha; STAT3: signal transducer and activator of transcription 3.
The LPS derived from bacteria reaches the liver through the portal circulation. It interacts with the TLR4 in the Kupffer calls, activating the NF-kB signaling cascade, leading to the production of proinflammatory cytokines, such as TNF-α and IL-1β41 (Fig. 1B). In chronic alcohol consumption, ethanol metabolism is increased, producing acetaldehyde and ROS that induce the activation of Kupffer cells, which trigger the production of a proinflammatory environment (TNF-α, IL-1, IL-6, IL-8, MCP-1, and TGF-β) and ROS, such as the superoxide anion, metabolites of arachidonic acid (prostaglandin D2, thromboxane A2, prostaglandin E2, and leukotrienes), favoring hepatocellular injury and/or death.36
A positive correlation between serum TNF-α levels with the Child-Pugh grading system has been described in patients with alcohol-induced cirrhosis.77 In a Sprague-Dawley mouse murine model, in which the animals were intraperitoneally given 1 µg of LPS from Escherichia coli per kilogram of body weight, TNF-α was reported to downregulate the expression of the PPARγ gene, favoring the expression of genes that participate in β-oxidation, contributing to the steatosis of ALD78 (Fig. 1C).
IL-6 is another cytokine that has been described in the context of ALD. It is a pleiotropic cytokine that has contrasting functions: it acts as a proinflammatory cytokine in chronic inflammatory disease models, and to the contrary, exhibits anti-inflammatory effects in acute inflammation, and has even been suggested to participate in liver regeneration phenomena79,80 (Fig. 1D and E). IL-6 has also been posited to have a protective effect in the early phase of ALD by participating in the suppression of chemokines that mainly attract neutrophils and monocytes.81–83 In addition, it can promote Th17 differentiation and IL-17 production, contributing to ethanol-induced inflammation of the liver84 (Fig. 1F). Increased IL-6 levels have been correlated with the chronicity and severity of ALD.85 IL-6 can protect against hepatocyte apoptosis and participate in mitochondrial DNA repair after an alcoholic liver lesion.86,87 (Fig. 1G). It is important to determine the stages, concentrations, and the molecular microenvironment that trigger either of the roles IL-6 can have during ALD.
On the other hand, in primary cultures of hepatocytes from rats that were chronically fed ethanol, there was increased cytotoxicity by TNF-α, as well as susceptibility to mitochondrial injury, triggering the processes of apoptosis and necrosis.88 Likewise, TNF-α produced by the Kupffer cells has been identified in those primary hepatocytes to induce the transcription of IL-8 mRNA89 (Fig. 1H).
IL-8, or CXCL-8, is one of the chemokines that has increasingly been accepted as an indicator of liver injury in ALD. That chemokine is involved in the mobilization of neutrophils from the bone marrow to the infiltration and activation of tissues90 (Fig. 1H). IL-8/CXCL-8 is produced by Kupffer cells in response to TNF-α and toll-like receptor (TLR) ligands through NF-kB activation.91 Activated HSCs secrete IL-8/CXCL-8 in response to stimulation by inflammatory cytokines, such as those produced by the Kupffer cells and some lymphocytes91 (Fig. 1H). In patients with AH, the expression of those chemokines in the liver is positively correlated with the severity of portal hypertension and patient survival.90,92
In animal models, as well as in patients with ALD, pro-IL-1β levels increase significantly in the liver and serum, compared with controls.93,94 In ALD, IL-1β participates in the recruitment and activation of NKT cells and neutrophils, causing inflammation and liver injury.95 That cytokine induces TGF-β expression by the Kupffer cells, promoting the activation of HSCs; the activated HSCs begin to produce the components of the extracellular matrix that, in a deregulated manner, leads to the progression of fibrotic events94 (Fig. 1I).
IL-4 is a pro-fibrotic mediator almost two times more efficacious than TGF-β for mediating fibrosis.96 It has inhibitory effects on IFN-γ.77 IL-13 is considered essential for the development of fibrosis because it stimulates the synthesis of collagen by fibroblasts and promotes the production of TGF-β.97 Both cytokines (IL-4, IL-13) have the same receptor in the fibroblasts that produce extracellular matrix proteins, such as type 1 collagen, type 3 collagen, and fibronectin, upon being stimulated through those interleukins.96,98,99
IL-10 is a cytokine that negatively regulates inflammatory responses through many mechanisms.100 An anti-inflammatory effect has been described in patients with fibrosis, upon promoting the apoptosis of macrophages with an inflammatory profile101 (Fig. 1J). In addition, it suppresses the synthesis of extracellular matrix proteins by fibroblasts, indicating a possible inhibition of fibrosis.45Table 1 describes the producer cells, target cells, and cellular effects, as well as the study models of cytokines and other cell mediators.
Cytokines that participate in ALD.
| Cytokine | Producer cell | Target cell | Receptor | Function and experimental and/or clinical evidence | ALD stage of cytokine participation | Study model | Ref |
|---|---|---|---|---|---|---|---|
| TNF-α | Kupffer cells | Kupffer cells | TNFR1 | Induction of IL-8 mRNA. | Inflammation | Liver tissueH | Thornton et al.89 |
| Hepatocytes | Hepatocytes | Induction of IL-6 and IL-1β via NF-kB. | Steatohepatitis | HepatocytesR | Pastorino et al.88 | ||
| Sinusoidal endothelial cells | HSCs | Apoptosis through p38MAPK, activating caspase-3, depolarization, and promoting programmed cell death. | Fibrosis | HSCsM | Saile et al.102 | ||
| HSCs | HSCs | Antiapoptotic and antiproliferative effect on activated HSCs. | Fibrosis (F1)* | Liver tissueM | Pradere et al.59 | ||
| Promotes HSC transdifferentiation into fibrogenic myofibroblasts via NF-kB. | Fibrosis (F1)* | HSCsMH | Tarrats et al.103 | ||||
| MMP-9 production due to TIMP1 mRNA reduction. | |||||||
| Extracellular matrix remodeling. | |||||||
| IL-1β | Kupffer cells | Kupffer cells | IL-1R1 | Promotes steatosis and liver injury. | Steatosis | Liver tissueM | Petrasek et al.94 |
| Promotes the expression of the inflammasome components pro-Casp-1, Asc, and Nlrp3. | |||||||
| Kupffer cell and lymphocyte recruitment and activation, favoring TNF-α, IL-6, MCP-1/CCL-2, and IL-10 production. | |||||||
| HSCs | Deficient activation of IL-1β via Casp-1, results in a decrease in liver fibrosis as well as in TGF-β1 and pro-Collal (fibrogenesis phenotype) expression. | ||||||
| Promotes the increase in the procollagen III N-terminal propeptide, TIMP-1, and hyaluronic acid. | Fibrosis (F1)* | Liver tissueM | Petrasek et al.94 | ||||
| SerumM | |||||||
| IL-6 | Kupffer cells | Kupffer cells | IL-6Ra | IL-6 deficiency results in the development of steatosis and high levels of malondialdehyde. | Steatosis | Liver tissueM | El-Assal et al.104 |
| Stromal cells | Kupffer cells | Upon receiving an exogenous dose of IL-6, reversal of the steatosis phenotype is seen. | |||||
| Dendritic cells | HSCs | sIL-6Ra binds to gp-130 | Promotes inflammation: | Steatosis | Liver tissueM | Naseem et al.105 | |
| -Transsignalling (soluble IL-6Ra). | |||||||
| -Proliferation and survival of T lymphocytes by means of IL-2 via STAT3/Bcl-2. | |||||||
| -Promotes CD8 T lymphocyte cytotoxicity. | |||||||
| -Differentiation into Th1 and Th17 lymphocytes. | |||||||
| Hepatoprotective role (regenerative and/or anti-inflammatory activity). | |||||||
| -Classic signaling (IL-6Ra). | |||||||
| -STAT3 and JAK activation. | |||||||
| -Strengthens angiotensinogen expression. | |||||||
| -Proliferation via MAPK and PI3K/Akt. | |||||||
| -Transition of the quiescent hepatocytes from phase G0 to G1 and S (regeneration). | |||||||
| -Reduces the apoptosis of hepatocytes through reduction of Bax and increase Bcl-1. | |||||||
| -DNA repair system activation through cell cycle inhibition. | |||||||
| -GreaterIRS-2 and G6Pase expression (glycogen storage). | |||||||
| -Antioxidant effect due to the increase in ATP production, metallothionine, and ROS suppression. -Differentiation to IL-22-producing T lymphocytes. | |||||||
| Fibroblasts | Sinusoidal cells | IL-6R | Cirrhosis | ||||
| Sinusoidal endothelial cells | Endothelial cells | IL-6 has a direct relation with the increase in C-reactive protein, IL-4, IL-10, and IFN-γ. | AH | SerumH | González-Reimers et al.77 | ||
| Mesenchymal cells | Hepatocytes | 40 mg/day of prednisone favors the hepatoprotective activity of IL-6 in up to 21% of patients treated, due to its increase. | |||||
| Higher IL-6 values are reported in patients with cirrhosis of the liver vs the control group. | Cirrhosis | Peripheral bloodH | Soresi et al.106 | ||||
| IL-8/CXCL8 | Kupffer cells | Hepatocytes | CXCR1 | Exacerbates lipid accumulation in the hepatocytes via Akt/HIF-1α. | Steatosis | Liver tissueM | Wang et al.45 |
| Activated HSCs | CXCR2 | Decreases PPARα expression, reducing β-oxidation. | |||||
| Damaged hepatocytes | Induces SREBP-1C activation, promoting adipogenic gene expression. | ||||||
| Neutrophils | Promotes neutrophil infiltration into the liver, activating these pathways: | AH | Liver tissueH | French et al.107 | |||
| -JAK2/STAT3 (proliferation), | NeutrophilsH | Takami et al.108 | |||||
| -PI3K/Akt (chemotaxis) | |||||||
| -MAPK (chemotaxis) | |||||||
| -Phospholipase C (PLC)/Protein kinase C (PKC) (cell activation) | |||||||
| Monocytes | -Promotes macrophage infiltration into the liver. | AH | Liver tissueH | Zimmermann et al.109 | |||
| Cirrhosis | |||||||
| Greater IL-8 expression in liver tissue is associated with poor outcome and neutrophil infiltration into the liver. Intrahepatic IL-8 expression is a predictor of mortality at 90 days. | AH | SerumH | Domínguez et al.90 | ||||
| Liver tissueH | |||||||
| IL-8 levels are high in Child C and F4, increasing in accordance with fibrosis grade and severity. | Cirrhosis | SerumH | Zimmermann et al.109 | ||||
| Liver tissueH | |||||||
| IL-10 | Kupffer cells | Kupffer cells | IL10R1 | Anti-inflammatory effect. | Fibrosis (F2) | Liver tissueMH | Wan et al.101 |
| Regulatory T lymphocytes | IL10R2 | Arginase activation in macrophages with an inflammatory profile (M1), promoting their apoptosis. | |||||
| Greater M1 macrophage apoptosis and M2 macrophage expression was detected in patients with liver damage. | Steatosis | Serum and liver tissueH | Wan et al.101 | ||||
| Fibrosis (F2) | |||||||
| IL-4 | Kupffer cells | Th2 lymphocytes | IL-4R | IL-4 levels progressively increase for 15 days in patients admitted with AH, possibly due to the change from the | AH | SerumH | González-Reimers et al.77; González-Reimers et al.110 |
| Th1 to Th2 response. | |||||||
| Low IL-4 levels in cirrhotic patients vs noncirrhotic patients and control group. | Cirrhosis | SerumH | González-Reimers et al.77 | ||||
| IL-13 | Lymphocytes (Th2) | Myofibroblasts | IL-4Rα-IL13Rα1 | Collagen synthesis is stimulated through the STAT6 pathway. | Fibrosis | Liver tissueM | Lee et al.111 |
| Kupffer cells | Induces TGF-β1 production. | Fibrosis | Liver tissueM | Lee et al.111 | |||
| Higher serum IL-13 levels in alcoholic subjects vs nondrinkers. | Cirrhosis | SerumH | González-Reimers et al.77 | ||||
| Higher levels in cirrhotics vs noncirrhotics, increasing in accordance with Child-Pugh class. | |||||||
| IFN-γ | NK | Hepatocytes | IFNGR | Attenuates steatosis, reducing lipogenesis. | Steatosis | Liver tissue and hepatocytesM | Cui et al.112 |
| Lymphocytes (Th1) | Kupffer cells | Promotes M1 macrophage activation, increasing their nitric oxide production and TNF-α expression. | Steatohepatitis | Monocytes/macrophagesM | Luo et al.113 | ||
| NKT | Hepatocytes | Apoptosis activation via JAK-STAT1-IRF3. | of all the subjects included in the study: | Liver tissueH | Stärkel et al.114 | ||
| Fibrosis | |||||||
| F1 (48%), | |||||||
| F2 (17%), | |||||||
| F3 (18%) | |||||||
| Cirrhosis (17%) | |||||||
| HSCs | Suppresses collagen synthesis through Smad7, attenuating TGF-β/SMAD3 signaling. | Fibrosis (F1)* | Liver tissueM | Sun et al.115 | |||
| HSCs isolated from liver tissueM | Jeong et al.116 | ||||||
| HSCs | Directly induces apoptosis of HSCs and arrests the cell cycle. Directly improves the cytotoxicity of the NKs toward the HSCs, through the TRAIL mechanism and NKG2D and through the IFN-γ/STAT1 signaling in the HSCs. | Cirrhosis | Liver tissueM | Radaeva et al.117 | |||
| NKs | Alcohol functions as an inhibitor of that pathway. | Lemmers et al.91 | |||||
| Alcohol induces greater una mayor expresión de TGF-β1expression, which inhibits the NKs through negative NKG2D, TRAIL, and IFN-γ regulation. | Fertin et al.96 | ||||||
| Cirrhotic patients have higher levels vs alcoholics with active consumption and no cirrhosis and a control group. | Alcoholics without ALD | SerumH | González-Reimers et al.77 | ||||
| Cirrhosis | |||||||
| IL-17 | Lymphocytes (Th17) | Neutrophils | IL-17RA | Promotes the infiltration of neutrophils into the liver. | Steatohepatitis | Liver tissueH | Lemmers et al.91 |
| HSCs | Activates JAK/STAT3; PI3K/Akt (proliferation and chemotaxis of neutrophils). | AH | |||||
| Stimulates IL-8 and CXCL1 production in HSCs. | Cirrhosis | ||||||
| HSCs | Promotes the profibrogenic process. | Fibrosis (F1)* | Liver tissueM | Meng et al.118 | |||
| Kupffer cells | Collagen 1 production in HSCs through STAT3 activation. | HSCs and Kuppfer cells isolated from liver tissueM | |||||
| TGF-β1 production by Kupffer cells through the NF-κB pathway. | |||||||
| Higher expression in F4 fibrosis vs initial and intermediate stages of fibrosis. | AH | Liver tissueH | Lemmers et al.91 | ||||
| Cirrhosis | |||||||
| TGF-β1 | Kupffer cells | HSCs | TGF-β1R | TGF-β2R phosphorilizes to TGF-β1R and activates SMAD2/3 stimulating the synthesis of extracellular matrix proteins, such as collagen I and II, and inhibiting their degradation. | Fibrosis (F1)* | Liver tissueM | Jeong et al.56; Qu et al.119; Zhang et al.120 |
| HSCs | NKs | TGF-β2R | Negatively regulates miRNA-29, producing positive regulation of the extracellular matrix proteins. Inhibits the expression of several antioxidant enzymes. | Fibrosis | Liver tissueM | Jeong et al.56; Qu et al.119; Zhang et al.120 | |
| Negatively regulates NKG2D, TRAIL, and IFN-γ expression, affecting the cytotoxicity of NKs toward HSCs. | |||||||
| MCP-1 or CCL2 | Kupffer cells | Circulatingmonocytes | CCR2 | Promotes hepatic circulating monocyte infiltration. | Inflammation | Circulating monocytesM | Mandrekar et al.121 |
| Hepatocytes | Kupffer cells | Induction of proinflammatory cyotokine mRNA expression: TNF-α, IL-1β, and IL-6. | Steatohepatitis | Liver tissueM | Mandrekar et al.121 | ||
| Monocytes | Induction of the mRNA expression of the ICAM adhesion molecules and CD68. | Inflammation | Liver tissueM | Mandrekar et al.121 | |||
| Sinusoidal endothelial cells | -Decrease in mRNA PPARα expression. | Steatohepatitis | HepatocytesM | Mandrekar et al.121 | |||
| Hepatocytes | -Decrease in binding of PPARα to DNA. | AH | SerumH | Fisher et al.122 | |||
| -Decrease in RNAm of ACC and CPT1. | Steatosis | ||||||
| Higher concentration in patients with AH vs. patients with cirrhosis and control subjects. | AH |
ACC: acetyl-CoA carboxylase; AH: alcoholic hepatitis; AKT: v-akt murine thymoma viral oncogene homolog; ALD: alcoholic liver disease; ASC: apoptosis-associated speck-like protein containing a CARD; ATP: adenosine triphosphate; BCL-1: B-cell lymphoma 1; BCL-2: B-cell lymphoma 2; CCR2: chemokine receptor 2; CPT1: carnitine palmitoyltransferase 1; CXCL-8: interleukin 8; CXCR1: CXC motif chemokine receptor 1; CXCR2: CXC motif chemokine receptor 2; DNA: deoxyribonucleic acid; G6Pase: glucose-6-phosphatase; HIF-1α: hypoxia-inducible factor 1-alpha; HSCs: hepatic stellate cells; ICAM: intercellular adhesion molecule; IFNGR: interferon gamma receptor; IFN-γ: interferon gamma; IL: interleukin; IL-1R1: interleukin 1 receptor type 1; IL-4R: interleukin 4 receptor; IL-4Rα/IL13Rα1: interleukin 4 receptor alpha/interleukin 13 receptor alpha 1; IL-6R: interleukin-6 receptor; IL-6Ra: interleukin-6 receptor subunit alpha; IL-10R1: interleukin 10 receptor alpha subunit; IL-10R2: interleukin 10 receptor beta subunit; IL-17RA: interleukin 17 receptor A; IRF3: interferon regulatory factor 3; IRS-2: insulin receptor substrate 2; JAK: Janus kinase, MCP-1: monocyte chemoattractant protein-1; MMP-9: metalloproteinase 9; mRNA: messenger RNA; NF-kB: nuclear factor-kappa B; NK: natural killer cells; NKG2D: natural killer group 2D receptor; NKT: natural killer T cells; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; PI3K: phosphoinositide 3-kinase; PPARα: peroxisome proliferator-activated receptor-alpha; p38 MAPK: p38 mitogen-activated protein kinase; ROS: reactive oxygen species; sIL-6Ra: soluble interleukin-6 receptor alpha; SMAD: Smad family; SREBP1: sterol regulatory element-binding protein 1; STAT3: signal transducer and activator of transcription 3; STAT6: signal transducer and activator of transcription 6; TGF-β1: transforming growth factor beta-1; TGF-β1R: transforming growth factor beta 1 receptor; TGF-β2R: transforming growth factor beta 2 receptor; TIMP1: tissue inhibitor of matrix metalloproteinase 1; TNFα: tumor necrosis factor-α; TNFR1: tumor necrosis factor receptor 1; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand.
Cell regulation also plays a dominant role in ALD. Such is the case of the NK cells, which have a key role in the defense of the host against viral infection, tumor transformation, and liver fibrosis inhibition. However, they contribute to the pathogenesis of inflammation and hepatic injury through their uncontrolled natural cytotoxicity and cytokine production.123,124 Activated NK cells produce cytokines, such as IFN-γ, TNF-α, IL-10, IL-12, and IL-22, and chemokines that include MIP-1α, MIP-β, RANTES, and IL-8.125 Through that mechanism, NK cells promote liver inflammation, culminating in liver injury. That damage is partially due to the expression of activating and inhibitory receptors in the NK cells and their ligands in hepatocytes and non-parenchymal cells, which are significantly altered during liver disease.126 Thus, the cytotoxic and proinflammatory activity of the NK cells against said cells is increased, resulting in the elimination of hepatocytes, and in turn, greater liver injury127,128 (Fig. 1K).
Immunologic regulation during liver fibrosisThe activation of HSCs is the key event in liver fibrogenesis1 (Fig. 1). Immunosuppression has been described as an important permissive factor for the development of fibrosis, suggesting that the immunologic state of the host regulates the progression of liver fibrosis.117,129,130 The much faster progression of liver fibrosis in patients with alcohol abuse and immunodepression is well documented.117,129,130 In addition, activated HSCs produce tissue inhibitors of metalloproteinase that attenuate metalloproteinase activity.131,132 Furthermore, HSCs are a principal source of TGF-β, which is the most potent inhibitor of NK cell functions, through the downregulation of the NKG2D receptor activator.56,123,133
NK cells and Th1 produce IFN-γ, which directly induces apoptosis and HSC cell cycle arrest.116,124 In addition, IFN-γ potentiates the cytotoxic activity of the NK cells, significantly contributing to their antifibrotic effect117 (Table 1). Alcohol consumption increases the cytotoxic activity of the NK cells in individuals that do not have ALD, contributing to the development of liver injury due to alcohol134 (Fig. 1K).
Our research group is currently carrying out studies directed at the changes and/or alterations of the different cell strains of the immune response and of various cell mediators at the peripheral level, to describe the cell changes and signaling pathways that participate in the different consumption patterns in AH and alcoholic liver cirrhosis.
ConclusionCytokines are a key piece in the pathogeny of ALD because they give rise to the perpetuation of the inflammatory state and liver injury. Likewise, it is important to emphasize that immunologic deregulation is a systemic process. Nevertheless, cytokines are also responsible for hepatoprotective mechanisms for preserving liver functionality. Thus, it is vastly important to know their functions in the development of ALD and create interventions that stop its progression and improve outcome. Despite the classification of ALD in stages, said stages are not separate, given that they can coexist in the same individual. It is crucial not to underestimate the consumption of alcohol. Alcohol abuse and dependence are not usually diagnosed, making it impossible to stop the progression of ALD.
PerspectivesALD is highly prevalent in Mexico, and so it is urgent to take prevention and treatment measures. Even though molecules that are useful as therapeutic targets or liver injury markers have already been identified, emphasis needs to be placed on the key mediators of the pathologic state of individuals with ALD. Having an understanding of the mechanisms involved in the pathogeny is the first step toward the development of therapeutic measures that can have an impact on the course of the disease. Likewise, the use of serum markers and other noninvasive methods could enable the identification of the disease at earlier stages, with the aim of having management regimens for patients with ALD. This would potentially reduce morbidity and mortality, given that at present transplantation is the only effective treatment, as well as ease the economic burden the disease produces on the national health system.
Ethical considerationsThe present work is a review manuscript and so does not involve patients, experimental animals, nor is it a clinical study. All information is cited correctly, respecting the authorship of each work. In addition, the research group and their experimental projects referring to alcoholic liver disease are carried out in accordance with the research commission and ethics committee of the Hospital General de México “Dr. Eduardo Liceaga” and the School of Medicine, UNAM.
Financial disclosureThe present study was partially funded by the Consejo Nacional de Ciencia y Tecnología (CONACyT), number SALUD-2016-272579, and the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-UNAM), number TA200515.
Conflict of interestThe authors declare there is no conflict of interest.
Please cite this article as: Martinez-Castillo M, Altamirano-Mendoza I, Sánchez-Valle S, García-Islas L, Sánchez-Barragán M, Hernández-Santillán M, et al. Desregulación inmunológica y fisiopatología del consumo de alcohol y la enfermedad hepática alcohólica. Rev Gastroenterol Méx. 2023;88:136–154.

