
La colangitis biliar primaria (CBP) es una enfermedad colestásica autoinmune, asociada a la presencia de anticuerpos antimitocondriales (AMA), exhibe un número extenso de manifestaciones autoinmunes extrahepáticas y un subgrupo de pacientes manifiestan otras enfermedades autoinmunes cutáneas1.
Presentamos el caso de una mujer de 45 años diagnosticada de CBP con 10 años de evolución en estadio avanzado, con cirrosis evidenciada por ultrasonido y Child-Pugh C (11 puntos), recibió tratamiento con ácido ursodeoxicólico con respuesta bioquímica adecuada, de acuerdo a los criterios de París hasta los 43 años, con incremento progresivo de AST, ALT, bilirrubinas y fosfatasa alcalina, y progresión de la enfermedad cambiando de Child-Pugh (A: 6 puntos; C: 11 puntos), liquen plano en la niñez y cáncer de mama diagnosticado en 2002, tratado con mastectomía radical, quimioterapia y radioterapia; en remisión total desde el 2008.
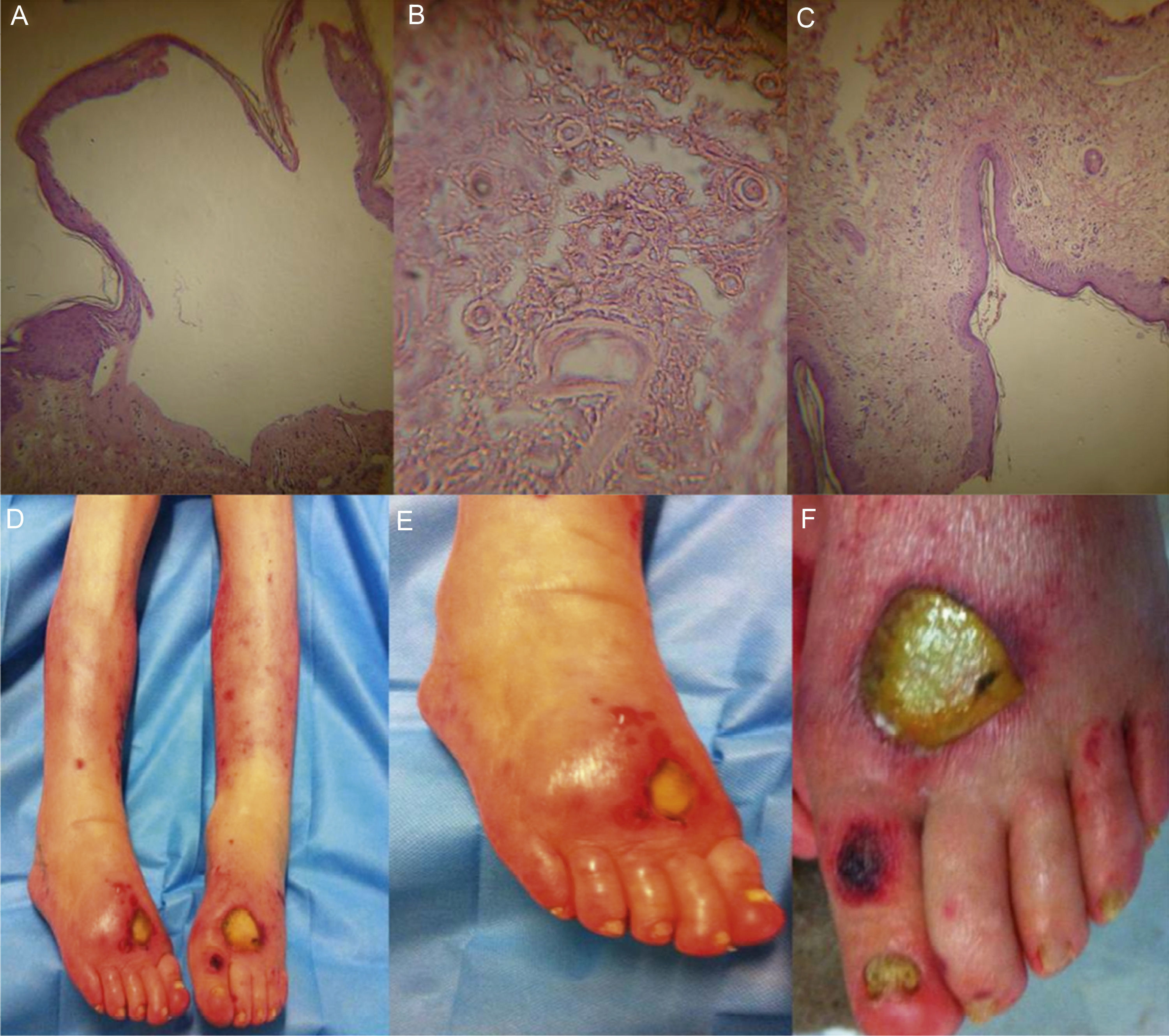
Acudió a urgencias por dermatosis de 21 días de evolución, caracterizada por lesiones vesiculares ampollosas frágiles con halo eritematoso. Inicialmente aparecieron en miembro torácico derecho, desapareciendo espontáneamente. Posteriormente se generalizaron a ambos miembros pélvicos. Al momento de la exploración dermatológica se observaron ámpulas de gran tamaño, denudadas con base cubierta con fibrina (fig. 1), algunas con borde necrótico, signo de Nikolsky negativo y ausencia de involucro a mucosas. Además, paroniquia, hiperqueratosis subungueal compatible con el diagnóstico de tiña pedis. Presentaba linfedema en miembro torácico derecho y mastectomía ipsilateral. La biopsia cutánea reportó ampolla subepidérmica con un infiltrado inflamatorio perivascular superficial mixto, compatible con pénfigo bulloso (PB), las lesiones cutáneas no fueron cultivadas, en la biopsia no se encontraron inclusiones virales. Durante la hospitalización la paciente cursó con infección de vías urinarias asociada a catéter vesical, lesión renal aguda, peritonitis bacteriana espontánea, seguida de choque séptico atribuido al foco urinario, que culminó en su deceso a los 5 días de su ingreso hospitalario.
 Cortes histológicos en donde se puede observar una ampolla subepidérmica con un infiltrado inflamatorio perivascular superficial mixto, compatible con pénfigo bulloso; D-F) Corresponden a lesiones dermatológicas constituidas por ámpulas de gran tamaño, denudadas con base cubierta con fibrina, algunas con borde necrótico.")
A-C) Cortes histológicos en donde se puede observar una ampolla subepidérmica con un infiltrado inflamatorio perivascular superficial mixto, compatible con pénfigo bulloso; D-F) Corresponden a lesiones dermatológicas constituidas por ámpulas de gran tamaño, denudadas con base cubierta con fibrina, algunas con borde necrótico.
Las manifestaciones dermatológicas comúnmente asociadas con CBP son: lesiones xantomatosas inespecíficas y melanosis; menos frecuentemente liquen plano, esclerodermia, síndrome de CREST (calcinosis cutis, fenómeno de Raynaud, alteraciones de la motilidad esofágica, esclerodactilia, telangiectasias) y el PB solo figura en reportes de caso2.
El PB es un padecimiento autoinmune cutáneo frecuente, representa el 70% de las dermatosis autoinmunes primarias. Se caracteriza por la presencia de autoanticuerpos dirigidos contra las protei¿nas antigénicas BP180, también llamadas bullous pemphigoid antigen 2 (BPAG2) o colágeno XVII y el BP230 o BPAG1 de los hemidesmosomas ubicados en la membrana basal. Clínicamente se caracteriza por la aparición inicial de lesiones urticariformes o eccematosas muy pruriginosas, sobre las que pueden aparecer ampollas tensas, estas pueden alcanzar un tamaño grande y suelen tener un contenido seroso o hemorrágico. Se localizan principalmente en tronco y superficie flexora de extremidades, el diagnóstico es clínico, histológico e inmunológico3.
La asociación de PB con otras enfermedades autoinmunes ha sido referida con anterioridad, pero no se ha demostrado una vía etiopatogénica común4.
Durante los últimos 36 años han sido reportados 6 casos de PB y CBP en la literatura. Hasta el momento se considera su asociación como fortuita. El pronóstico de supervivencia de los pacientes con PB sin otros padecimientos autoinmunes es malo, alcanza el 30% a un año. La muerte ocurre frecuentemente dentro de las primeras 12 semanas del inicio del tratamiento. Las causas de muerte incluyen efectos adversos de la terapia, infecciones agregadas y complicaciones relacionadas a la enfermedad subyacente5.
Se desconoce el comportamiento habitual en pacientes con CBP y PB coincidente, los reportes previos6–8, hacen referencia a pacientes de reciente diagnóstico de CBP. Con respecto a la respuesta al tratamiento, existen reportes a corto plazo. A pesar de su rareza es importante iniciar un tratamiento oportuno y multidisciplinario, el tratamiento recomendado en la mayoría de las guías incluye prednisona (0,75mg/kg), dapsona (100mg/24h) o azatioprina (100-150mg/día). El objetivo es evitar complicaciones tales como extensión y severidad de la enfermedad, y desarrollo de infecciones que ensombrezcan el pronóstico, como en este caso.
El desafortunado caso de nuestra paciente refleja el retraso en la búsqueda de atención médica, lo que culminó en un desenlace fatal, por otro lado el inicio temprano de la enfermedad se asocia a un peor pronóstico, mayor tasa de fallos al tratamiento.
Finalmente, a pesar de que no se trata de un caso autoinmunitario múltiple, debido a lo inusual de la presentación, decidimos reportarlo para divulgar su asociación y aumentar así la sospecha clínica.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

