
Paciente varón de 18 años, que es remitido por los hallazgos de un estudio baritado donde se describe pólipo sésil de unos 15mm en íleon.
Como antecedentes familiares refiere que su padre ha sido intervenido por pólipos de colon y de intestino delgado sin llegar a ser estudiado. Entre los antecedentes personales destaca que a los 9 años presentó un episodio de hematoquecia secundario a pólipos de colon, que fueron resecados endoscópicamente, sin realizar seguimiento posterior.
El paciente está asintomático, sin dolor abdominal ni alteración del ritmo intestinal. A la exploración física se objetivan máculas milimétricas pigmentadas en el labio inferior y dedos de la mano, sin otras alteraciones.
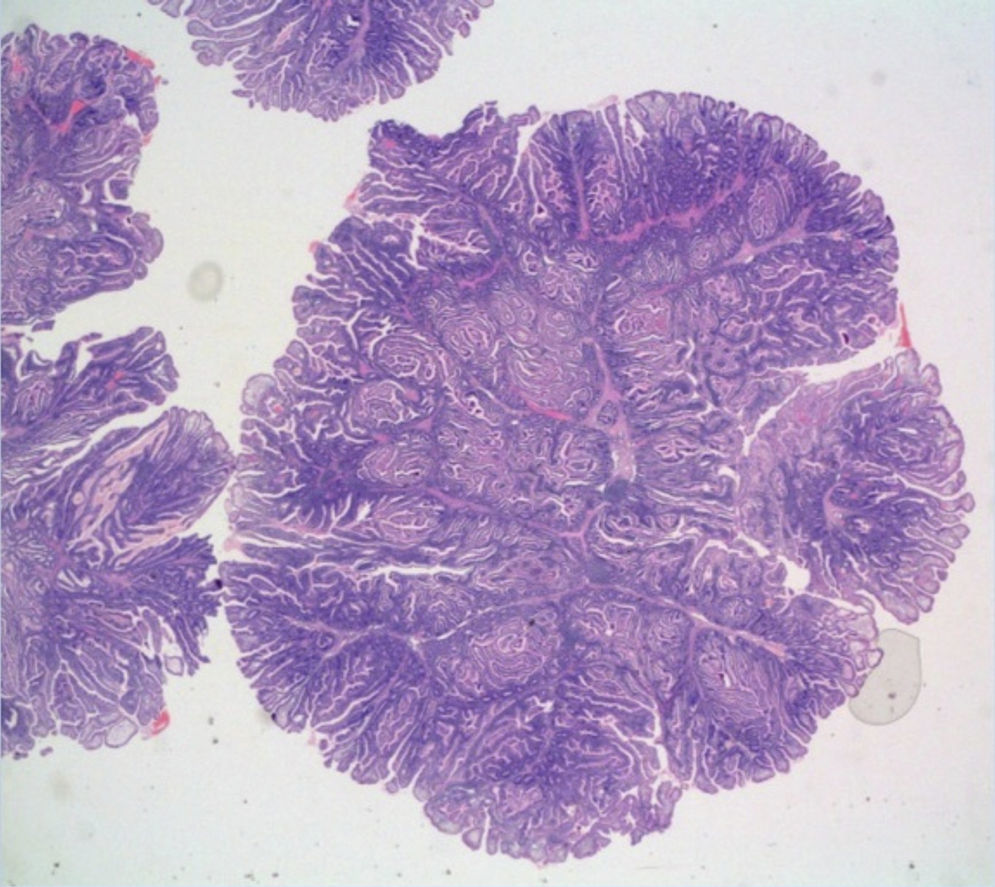
La lesión polipoidea se confirma mediante cápsula endoscópica e ileoscopia realizadas al mes, pero no es posible su exéresis endoscópica, por lo que se decide resección ileocólica con el hallazgo de 2 pólipos hamartomatosos sin displasia (fig. 1).

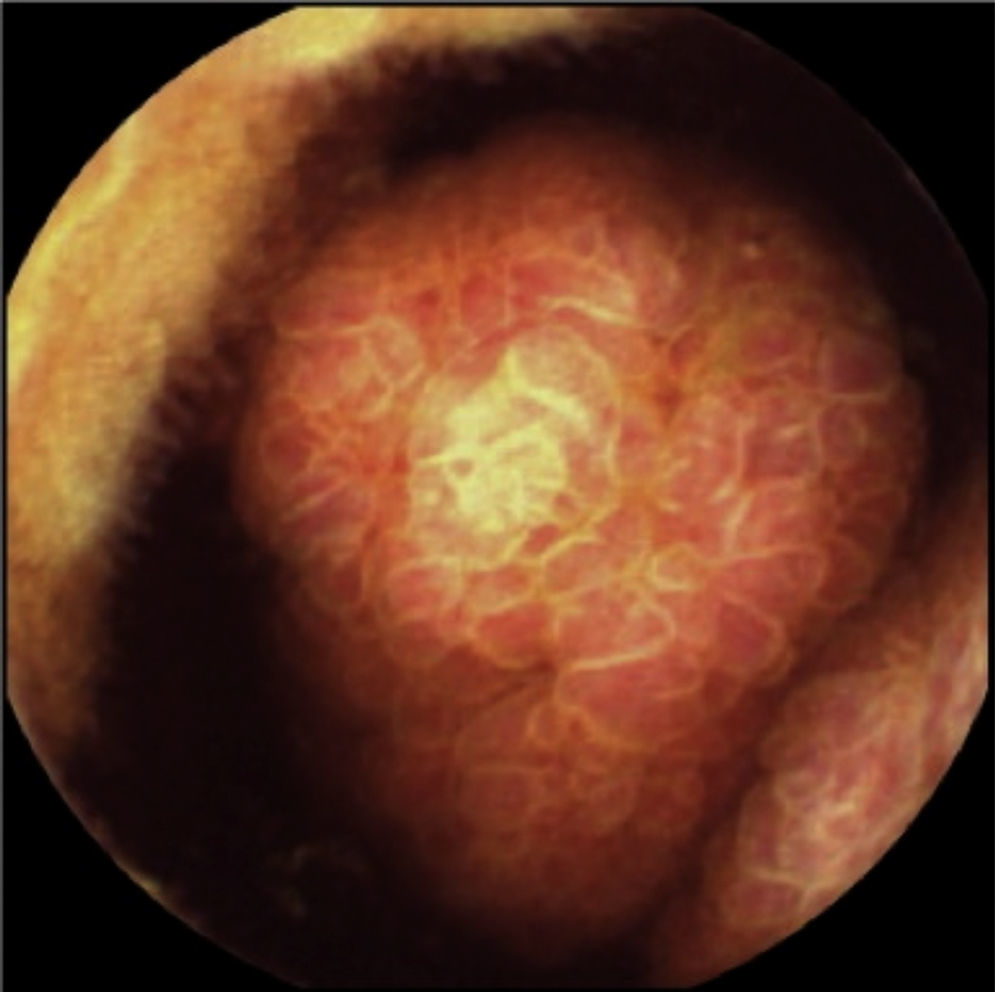
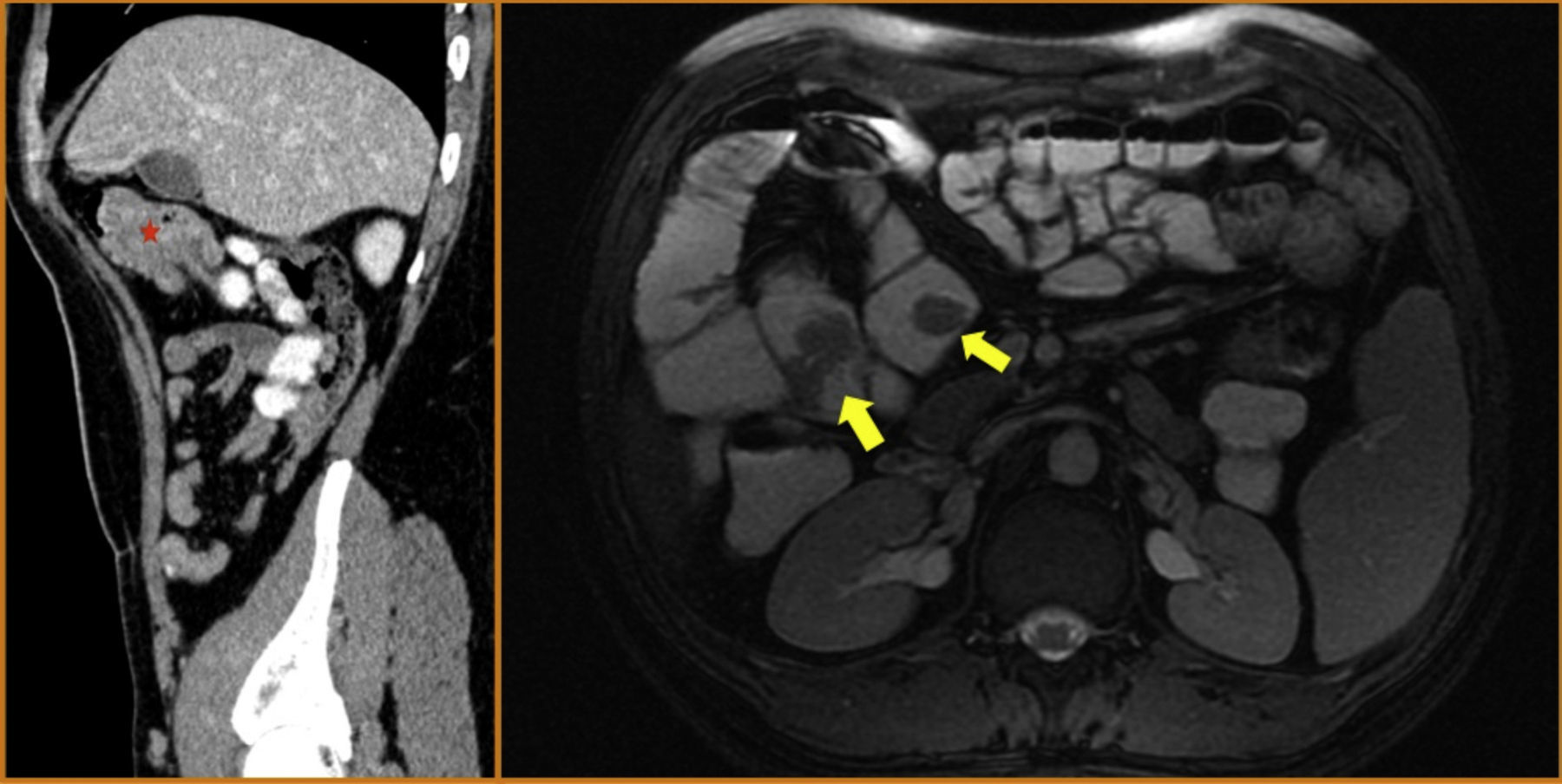
El paciente permanece asintomático durante el seguimiento con resección de múltiples pólipos colónicos y gástricos. Sin embargo, en la última cápsula endoscópica, realizada 2 años después de la cirugía, se describe un gran pólipo ulcerado en el yeyuno, que ocupa toda la luz intestinal (fig. 2). Estos hallazgos se confirman mediante entero-resonancia y tomografía axial computarizada en donde se objetivan varias lesiones polipoideas en yeyuno que están sirviendo como cabeza de invaginación de un segmento intestinal (fig. 3).

. A la derecha, imagen axial de entero-resonancia en la que se identifican varias lesiones polipoideas en yeyuno e íleon proximal (flechas amarillas).")
A la izquierda imagen sagital de TC en la que se evidencian varias formaciones polipoideas que están sirviendo de cabeza de invaginación de un segmento yeyunal (asterisco). A la derecha, imagen axial de entero-resonancia en la que se identifican varias lesiones polipoideas en yeyuno e íleon proximal (flechas amarillas).
Dos meses más tarde, tras intento fallido de llegar a la lesión mediante enteroscopia de pulsión, se realiza una enteroscopia de doble balón, en la que vía oral se alcanza el pólipo descrito, pero resulta imposible su resección endoscópica. Por ello, el paciente se somete al mes a una nueva cirugía en la que, a través de varias enterotomías, se resecan 10 pólipos hamartomatosos, algunos de los cuales incluidos en un segmento invaginado.
Ante estos datos clínicos e histopatológicos se llega al diagnóstico de síndrome de Peutz-Jeghers (SPJ). Se solicita estudio genético en el que, tras secuenciación completa del gen STK11 (LKB1), se confirma que el paciente es portador de una mutación patogénica en el exón 4 (p.Asp194Asn). Tras la cirugía, el paciente ha permanecido asintomático y sin hallazgos relevantes en las pruebas de vigilancia.
El SPJ es una enfermedad genética rara con herencia autosómica dominante, causada por una mutación germinal en el gen STK11 (LKB1) localizado en el cromosoma 19p13.3. Su incidencia se estima de uno por cada 50.000-200.000 nacimientos1. Precisamente debido a la baja frecuencia de este síndrome, no existen protocolos bien establecidos sobre su vigilancia y manejo.
El diagnóstico del SPJ se basa en la presencia de algunos de estos criterios2:
- −
Si no existen antecedentes familiares de SPJ:
- •
Dos o más pólipos hamartomatosos confirmados histológicamente, o
- •
Cualquier número de pólipos hamartomatosos con pigmentación mucocutánea característica
- •
- −
Si existen antecedentes familiares de SPJ, cualquier número de pólipos hamartomatosos o pigmentación mucocutánea
Las lesiones cutáneas aparecen en el 95% de los pacientes, y consisten en máculas pigmentadas de color marrón oscuro-negras cuya localización más frecuente es en la región perioral (94%), manos (74%) y mucosa bucal (66%)3.
La gran mayoría de los pacientes va a presentar pólipos hamartomatosos. Se suelen localizar con más frecuencia en intestino delgado (60-90%), seguido de colon (50-64%), estómago (49%) y recto (32%), pudiendo incluso ser extraintestinales. El número y tamaño es muy variable y endoscópicamente no tienen características específicas4.
Para realizar un adecuado diagnóstico diferencial con el resto de poliposis debemos conocer que existen 3 tipos de pólipos, cada uno de los cuales se asocia a un síndrome determinado. Dentro de los adenomas, la poliposis adenomatosa familiar clásica o atenuada o la poliposis asociada al gen MYH son los síndromes más frecuentes. Asociado a los pólipos serrados existe la poliposis serrada. Finalmente, dentro de la poliposis hamartomatosas, además del SPJ, se describe la poliposis juvenil, el síndrome de Cowden y el síndrome de Bannayan-Riley- Ruvalcaba, con manifestaciones clínicas y extraintestinales diferentes5.
La clínica más frecuente de comienzo en el SPJ es la obstrucción intestinal (43%), seguido de dolor abdominal de origen isquémico (23%), rectorragia (14%) o extrusión del pólipo por el recto (7%)1. Hasta un 69% de los pacientes puede presentar algún fenómeno de invaginación producida por pólipos mayores de 15mm, por lo que la resección de pólipos de estos tamaños podrían evitar dichos cuadros6.
En estos pacientes existe, además, un riesgo incrementado de tumores. Las neoplasias más frecuentes son las de colon, seguido de mama y de intestino delgado. El riesgo acumulativo aumenta con la edad, y varía del 37 al 93% con un riesgo relativo de 9,9-187.
El riesgo de invaginación y de neoplasias es lo que debe vigilar en estos pacientes. Sin embargo, ningún protocolo de cribado ha sido validado en ensayos clínicos. Existe una guía en la que se establecen unas recomendaciones que incluye no solo la vigilancia endoscópica de pólipos gastrointestinales, si no también el cribado de neoplasias extraintestinales como el cáncer de mama o testicular8. Recientemente, el grupo internacional para el cribado de cáncer de páncreas ha incluido dentro de los pacientes con alto riesgo a los pacientes con SPJ, independientemente de la historia familiar de cáncer de páncreas9.
No existe consenso sobre cómo tratar los pólipos cuando se detectan, pero, aunque la detección de displasia en los hamartomas sea baja, la polipectomía endoscópica es segura, ya que evita la cirugía de urgencia y protege del desarrollo de neoplasias intestinales y extraintestinales10.
FinanciamientoNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.

