




¿ Conceptos clave:
El hígado graso no alcohólico (HGNA) es una entidad con una prevalencia mundial cercana a 20% a 30%, que abarca de la esteatosis simple a su manifestación inflamatoria (esteatohepatitis), la cual puede evolucionar a fibrosis, cirrosis y en contadas ocasiones a carcinoma hepatocelular.
Representa el componente hepático del síndrome metabólico que incluye resistencia a la insulina, dislipidemia, obesidad (central) e hipertensión arterial sistémica.
El diagnóstico definitivo es histológico con esteatosis macrovesicular como hallazgo característico. También se puede observar infiltrado inflamatorio mixto, núcleos glucogenados, degeneración ba lonoide, cuerpos hialinos de Mallory y, en algunos casos, grados variables de fibrosis.
La terapia farmacológica mejora la esteatosis y la inflamación; no hay datos que soporten una mejoría en el grado de fibrosis.
¿ Perspectiva histórica
En 1980 Ludwig y colaboradores acuñaron el término de esteatohepatitis no alcohólica (EHNA) para describir una serie de 20 pacientes evaluados en la Clínica Mayo a lo largo de diez años, quienes tenían evidencia histológica de hepatitis alcohólica sin historia de abuso de alcohol.1 Ludwig dividió a la EHNA en dos grupos: EHNA primaria asociada a la obesidad y EHNA secundaria, la cual contempla complicaciones de cirugía bariátrica, fármacos, o se asocia con otras condiciones como la enfermedad de Wilson y la abetalipoproteinemia.
¿ Epidemiología del hígado graso no alcohólico
A pesar de que hacen falta más estudios epidemiológicos, el HGNA es la enfermedad hepática más frecuente en el mundo y la causa más común de alteraciones en las pruebas de funcionamiento hepático en Estados Unidos.2,3 El HGNA afecta tanto a niños como a adultos y su prevalencia en la población general se estima entre 2.8 y 24%.4,5 En México estudios poblacionales han estimado una prevalencia de alrededor de 17.05% en población asintomática.6 En la serie de 351 autopsias de pacientes obesos y no obesos publicada por Wanless y colaboradores se encontró esteatosis hepática en el 70% de los primeros y en el 35% de los no obesos. La prevalencia de EHNA fue de 6.5%.7 Diversos estudios muestran que en Estados Unidos la prevalencia de HGNA es mayor en la población hispana y méxico-americana. La Tabla 1 muestra las condiciones que con más frecuencia se asocian a HGNA. La prevalencia y severidad del HGNA se correlacionan con el grado de obesidad. En diversos estudios se ha demostrado una relación entre el índice de masa corporal, el grado de esteatosis, y la gravedad de la lesión hepática. Sin embargo, la distribución de la grasa corporal parece ser más importante en el desarrollo de esteatosis que la masa adiposa total.8,9 La gran asociación entre la obesidad y el HGNA, aunada al rápido incremento en la prevalencia mundial de la obesidad sugiere que la prevalencia del HGNA continuará en aumento.

El HGNA igualmente se asocia a diabetes mellitus tipo 2 (DM2) y a intolerancia a la glucosa con o sin obesidad. Alrededor del 20% al 70% de los pacientes adultos con EHNA tienen DM2, hiperglucemia o intolerancia a la glucosa.8,10 Se dispone de evidencia que respalda la asociación de la resistencia a la insulina y la hiperinsulinemia, aún en sujetos con tolerancia normal a la glucosa.11,12 La DM2 ha mostrado ser un factor predictor independiente de enfermedad hepática avanzada en pacientes con HGNA, y es un factor de riesgo independiente para cirrosis y carcinoma hepatocelular. El riesgo y la gravedad del HGNA aumentan con el número de componentes del síndrome metabólico.13 La dislipidemia es un hallazgo frecuente; se observa hipertrigliceridemia, hipercolesterolemia o ambas hasta en 20% a 81% de los pacientes. La mayoría de los pacientes con HGNA tiene múltiples factores de riesgo incluyendo DM2 y dislipidemia.
¿ Fisiopatología
La esteatosis hepática se caracteriza por la acumulación de triglicéridos en macro y microvesículas en más del 5% de los hepatocitos, distribuyéndose principalmente en la región perivenular; las áreas periportales usualmente se encuentran respetadas.14
La esteatosis hepática se desarrolla como consecuencia de disfunción de diversas vías metabólicas. El incremento en la concentración de ácidos grasos libres (AGL) parece ser un factor determinante en la patogénesis del hígado graso. Sin embargo, recientemente se ha reconocido el rol desempeñado por la transcripción de ciertos factores, la acción de las adipocitocinas, y las alteraciones en la peroxidación lipídica.15
La resistencia a la insulina, presente en un gran porcentaje de pacientes, juega un papel importante en la patogénesis del HGNA.16,17 La esteatosis hepática grave se observa asociada a una gran resistencia a la insulina y altos niveles de AGL. Concentraciones elevadas de insulina ocasionan falla en la supresión del flujo de ácidos grasos en pacientes con esteatosis hepática.18 Existe una relación dosis-dependiente entre la acumulación intrahepática de grasa y la resistencia hepática a la insulina. Este proceso puede estar mediado por alteraciones en la fosforilación del receptor de insulina (IRS-1 y 2) y otras vías de señalización de la insulina (proteincinasa-B, proteincinasa C-b y la cinasa c-Jun N-terminal-1).19 En personas sin HGNA, la reserva de AGL en el tejido adiposo contribuye a la mayor parte del flujo de ácidos grasos al hígado durante el ayuno; sin embargo, para el HGNA son importantes otras fuentes de grasas como la lipogénesis de novo y los ácidos grasos procedentes de la dieta, los cuales pueden ingresar en el plasma como excedente o a través de la obtención de remanentes de quilomicrones. Evidencia reciente indica que durante la entrega aumentada de ácidos grasos a los hepatocitos, la lipogénesis de novo se incrementa en el HGNA como resultado de la sensibilidad preservada de las vías de lipogénesis a la acción de la insulina y la sobreexpresión de la proteína de unión al elemento de respuesta a los esteroles (SRBEP)-1c. SRBEP-1c es la mayor isoforma expresada en el hígado y en tejidos involucrados en la homeostasis energética, y es activada por la insulina, el receptor hepático X (LXR)-α, el receptor endocanabinoide CB1 y el supresor del señalamiento de citocinas (SOC)-3 e inhibido por el glucagon. En modelos murinos con sobreexpresión de SREBP-1c se observó el desarrollo de lipodistrofia, resistencia a la insulina y esteatosis hepática.20
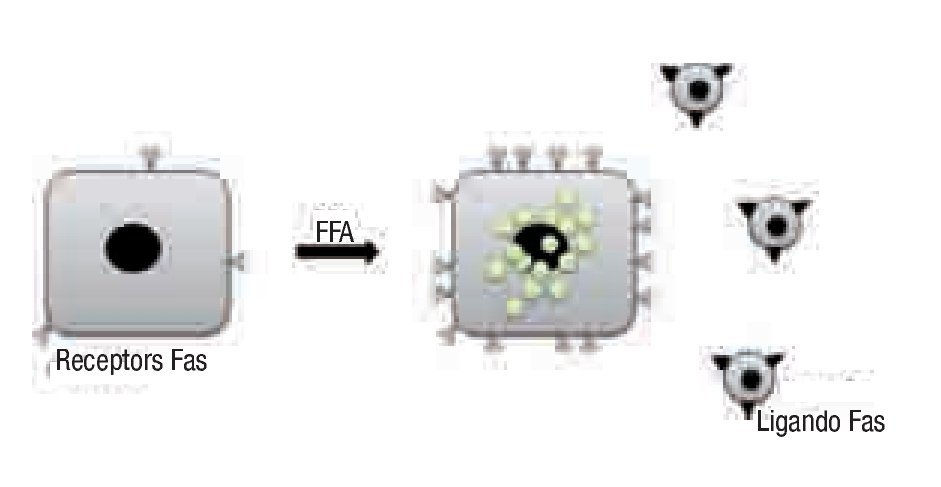
Por otra parte, se ha postulado que una de las maneras en que la grasa provoca lesión intrahepática es la regulación positiva de la molécula proapoptótica Fas (CD 95) (Figura 1). La expresión de Fas aumenta en el hígado de pacientes con EHNA, y se sugiere que juega un importante papel en la apoptosis que acompaña a la esteatohepatitis.21 Otros efectos tóxicos de los AGL son la liberación de proteasas lisosómicas como la catepsina B hacia el citosol,22 proceso que involucra la translocación de la molécula formadora de poros Bax a la membrana lisosomal (Figura 2).23

¿ Figura 1.Los ácidos grasos incrementan la expresión hepática de Fas. Los ácidos grasos libres en cultivo celular, así como la alimentación alta en calorías in vivo causan regulación positiva del receptor Fas en los hepatocitos. Esto coloca a las células en riesgo de muerte tanto por ataque por células inflamatorias que expresan ligando Fas (células T citotóxicas o células NK), como por señalización espontánea inducida por agregación de moléculas receptoras densamente expresadas en la superficie celular.21

¿ Figura 2.Los ácidos grasos libres causan desestabilización lisosomal que culmina en la producción hepática de TNF-a. En hepatocitos normales la catepsina B se localiza en los lisosomas y Bax se distribuye por todo el citoplasma. Los ácidos grasos promueven la localización de Bax hacia las membranas lisosómicas lo que da lugar a desestabilización y salida de catepsina B al citoplasma. A su vez la catepsina B activa a NF-kB , el cual induce la expresión de TNF-a. El TNF-a causa muerte celular al amplificar la desestabilización lisosomal, y promueve la esteatosis hepatocelular.23
En los pacientes con EHNA la actividad del complejo de cadena respiratoria está reducida, como también lo está la capacidad para resintetizar adenosín trifosfato (ATP), con disminución del ATP hepático de forma transitoria. Las mitocondrias del hígado exhiben daño ultraestructural con presencia de inclusiones paracristalinas en megamitocondrias, con niveles disminuidos de ADN mitocondrial, reducción de la expresión proteica de diversos polipéptidos codificados por ADN mitocondrial, y menor actividad en los complejos I, III, IV y V (ATP sintetasa) codificados por ADN mitocondrial y complejo II codificado por ADN nuclear.24 -27 En presencia de resistencia a la insulina, el efecto normal de esta hormona sobre la peroxidación lipídica se encuentra reprimido. Como resultado de esto la b-oxidación mitocondrial de los ácidos grasos aumenta, lo que a su vez incrementa la formación de especies reactivas de oxígeno (ROS) debido a la ineficiencia en el acoplamiento de la oxidación con la fosforilación en la mitocondria. La generación de ROS en la mitocondria puede tener consecuencias adversas adicionales como el daño oxidativo en el ADN mitocondrial, aumento en la producción de TNF-α y lipoperoxidación directa de las membranas mitocondriales, lo cual produce pérdida del citocromo C de las mitocondrias.28,29 La depleción progresiva de citocromo C inicia la apoptosis y la muerte celular. Un segundo mecanismo para la generación de ROS es la activación del sistema de citocromo P450, cuyos componentes principales son CYP2E1 y CYP4A. En estados normales la insulina regula a la baja la expresión de CYP2E1, mientras que en la resistencia a la insulina se observa una expresión aumentada debido a la falta de los efectos represores de esta hormona. En los pacientes con EHNA, la actividad de CYP2E1 se encuentra persistentemente elevada con máxima distribución en la zona 3, el sitio con mayor afectación en esta condición patológica.30-32
¿ Histología
El espectro de las anormalidades histológicas encontradas en el HGNA varía desde la esteatosis simple hasta la cirrosis. El hígado graso simple se caracteriza por esteatosis macrovesicular de diversos grados, que usualmente es difusa pero ocasionalmente centrolobular. Puede haber alteraciones inflamatorias por linfocitos, neutrofilos o mixtos. No se observa fibrosis en la esteatosis simple. Comúnmente se observan núcleos glucogenados en esteatosis asociada a obesidad y diabetes mellitus.
El HGNA avanzado, conocido también como EHNA, histológicamente es indistinguible de la hepatitis alcohólica. Se han sugerido diversos criterios histológicos para establecer el diagnóstico de EHNA, los cuales aún no están bien definidos (Tabla 2).

La fibrosis es una de las principales características de la EHNA. Se ha descrito fibrosis pericelular, perisinusoidal y periportal en 37% a 84% de los pacientes con EHNA. La extensión de la fibrosis puede variar considerablemente, desde delgadas fibras alrededor de venas pequeñas o grupos de células, hasta densos septos fibróticos con distorsión de la arquitectura hepática. La fibrosis perisinusoidal es la más común, especialmente en adultos; inicialmente es leve y predomina en la zona 3 alrededor de las venas hepáticas. Puede haber cirrosis en la biopsia inicial en 7% a 16% de los pacientes con HGNA con alteración de las pruebas de función hepática. El riesgo de cirrosis puede ser mayor en los pacientes con obesidad mórbida.
¿ Estrategias diagnósticas
La mayoría de los pacientes con HGNA son evaluados por elevación crónica de las pruebas de funcionamiento hepático, hepatomegalia, o ambas. La combinación de historia, exploración física, pruebas sanguíneas no invasivas, y exámenes radiológicos es útil para excluir otras causas de enfermedad hepática.
Las pruebas de laboratorio deben incluir perfil hepático completo, biometría hemática, tiempo de protrombina, anticuerpos contra el virus de hepatitis C, antígeno de superficie de hepatitis B, pruebas de hierro, ceruloplasmina en pacientes jóvenes, anticuerpos antinucleares, α1-antitripsina y anticuerpos antimitocondriales. El ultrasonido hepático es la prueba de imagen más utilizada; usualmente revela un hígado brillante con ecogenicidad aumentada, aunque la ausencia de estos hallazgos no excluye el diagnóstico de HGNA.
Un aspecto importante en el establecimiento del diagnóstico de HGNA es poder distinguirlo de la enfermedad alcohólica. Histológicamente existen pocas diferencias entre la EHNA y la enfermedad hepática por alcohol. Es por eso que el diagnóstico de EHNA o esteatohepatitis alcohólica debe basarse en la ausencia de abuso de alcohol (< 20 a 40 g de alcohol al día). Es preciso obtener una historia detallada del consumo de alcohol interrogando al paciente y a miembros de la familia.
El papel de la biopsia hepática es controversial. Muchos médicos consideran al HGNA como un diagnóstico de exclusión cuando las pruebas clínicas y de laboratorio no revelan otra causa de daño hepático crónico, y el examen radiológico muestra hígado graso. En su mayoría, los pacientes con HGNA no son sometidos a biopsia. El mayor argumento en contra de la realización de la biopsia es que los hallazgos histológicos no afectarán el manejo de la mayoría de los pacientes, debido a que las opciones terapéuticas son pocas, y el grado de daño observado no modifica el manejo más allá de la reducción de peso y la realización de actividad física.
La tomografía computada puede detectar el contenido intrahepático de grasa pero únicamente en un umbral del 30%. La resonancia magnética es probablemente el método más certero y rápido para medir la grasa hepática; sin embargo, es costoso y el software necesario no está disponible en todas las unidades de resonancia magnética.33 Roldan Valadez y colaboradores34 en su estudio de cuantificación de grasa intrahepática por espectroscopía en 18 pacientes demostraron que con un valor de corte de 7.48% de grasa intrahepática, el diagnóstico se realizaba en 100% de los casos, mostrando que puede reemplazar a la biopsia hepática para este fin.
¿ Consideraciones generales para el tratamiento
Se deben considerar algunos aspectos en la toma decisiones terapéuticas en caso de HGNA. Primero, no se dispone de un tratamiento (farmacológico o quirúrgico) que muestre beneficio para la reducción de la fibrosis. Segundo, los únicos estudios controlados publicados han demostrado un efecto marginal en comparación con placebo. Tercero, existen retos importantes en el diseño y la realización de ensayos clínicos sobre HGNA. Cuarto, el HGNA es el componente hepático del síndrome metabólico y el tratamiento inicial de todas las formas de HGNA debe estar dirigido a mejorar las comorbilidades como obesidad central, dislipidemia, hipertensión y diabetes. La Tabla 3 muestra las estrategias terapéuticas aplicadas actualmente de acuerdo al componente fisiopatológico del HGNA.34-39

Por lo tanto, fuera de la reducción de peso y los cambios en la dieta, las restantes estrategias terapéuticas son consideradas experimentales y deben ser evaluadas bajo protocolos de investigación.
¿ Conclusión
El HGNA es una enfermedad de alta prevalencia, que se acompaña de las comorbilidades asociadas al síndrome metabólico. Los eventos fisiopatológicos involucrados se encuentran parcialmente descritos y se asocian con resistencia a la insulina y lipotoxicidad. Actualmente el manejo quirúrgico o farmacológico se recomienda únicamente dentro de ensayos clínicos controlados, por lo que la actividad física y las modificaciones de la dieta continúan siendo la base del manejo, aunque la tasa de apego a estas intervenciones es baja.

