


La deficiencia de lipasa ácida lisosomal (DLAL) ocasiona el almacenamiento de ésteres de colesterol y triglicéridos en los lisosomas de los hepatocitos y células del sistema monocito-macrófago y, como consecuencia, produce una enfermedad sistémica con manifestaciones variadas que puede pasar inadvertida; es indispensable reconocerla ya que puede diagnosticarse a cualquier edad y recibir tratamiento específico. El objetivo de este documento es ofrecer una guía que permita al médico conocer los aspectos fundamentales relacionados con el diagnóstico de la DLAL para garantizar su identificación. Este documento fue diseñado por un grupo de expertos y se presenta como un algoritmo para orientar al médico pediatra, internista, gastroenterólogo, endocrinólogo, genetista, patólogo, imagenólogo y otros especialistas que pudieran enfrentar a esta entidad.
MétodosSe realizó una revisión actualizada de la literatura con respecto a las manifestaciones clínicas y el diagnóstico de la DLAL por parte de los expertos mexicanos. Se plantearon las declaraciones con base en esta revisión y se sometieron a votación. Se utilizó el método cuantitativo estructurado de técnica de grupo nominal para alcanzar un consenso.
ResultadoSe propone un algoritmo práctico del proceso diagnóstico de pacientes con DLAL, con base en datos clínicos y de laboratorio indicativos de la enfermedad, acorde con el consenso estabilizador para cada recomendación.
ConclusiónEste algoritmo proporciona una secuencia de acciones clínicas, basado en las manifestaciones clínicas obtenidas de los diferentes estudios, con el propósito de optimizar el proceso diagnóstico de los pacientes con sospecha de DLAL.
Lysosomal acid lipase deficiency (LAL-D) causes progressive cholesteryl ester and triglyceride accumulation in the lysosomes of hepatocytes and monocyte-macrophage system cells, resulting in a systemic disease with various manifestations that may go unnoticed. It is indispensable to recognize the deficiency, which can present in patients at any age, so that specific treatment can be given. The aim of the present review was to offer a guide for physicians in understanding the fundamental diagnostic aspects of LAL-D, to successfully aid in its identification.
MethodsThe review was designed by a group of Mexican experts and is presented as an orienting algorithm for the pediatrician, internist, gastroenterologist, endocrinologist, geneticist, pathologist, radiologist, and other specialists that could come across this disease in their patients. An up-to-date review of the literature in relation to the clinical manifestations of LAL-D and its diagnosis was performed. The statements were formulated based on said review and were then voted upon. The structured quantitative method employed for reaching consensus was the nominal group technique.
ResultsA practical algorithm of the diagnostic process in LAL-D patients was proposed, based on clinical and laboratory data indicative of the disease and in accordance with the consensus established for each recommendation.
ConclusionThe algorithm provides a sequence of clinical actions from different studies for optimizing the diagnostic process of patients suspected of having LAL-D.
La deficiencia de lipasa ácida lisosomal (DLAL) es una enfermedad autosómica recesiva que se caracteriza por acumulación progresiva de ésteres de colesterol y triglicéridos en los lisosomas de los hepatocitos y del sistema monocito-macrófago, no es exclusiva de los niños pues se ha diagnosticado a cualquier edad; es una causa poco reconocida de dislipidemia asociada al desarrollo de aterosclerosis, enfermedad cardiovascular y enfermedad hepática progresiva. Característicamente se presenta como hepatomegalia, elevación de aminotransferasas y esteatosis microvesicular difusa en la biopsia hepática1,2.
La DLAL es una enfermedad que puede pasar inadvertida si no se piensa en ella, o confundirse con otras entidades como enfermedad por hígado graso no alcohólico (EHGNA), esteatosis hepática no alcohólica, hipercolesterolemia familiar heterocigota o hiperlipidemia familiar combinada, entre otras2.
El objetivo del presente trabajo es proponer una guía rápida para sospechar e identificar la DLAL; para ello se propone un algoritmo diagnóstico.
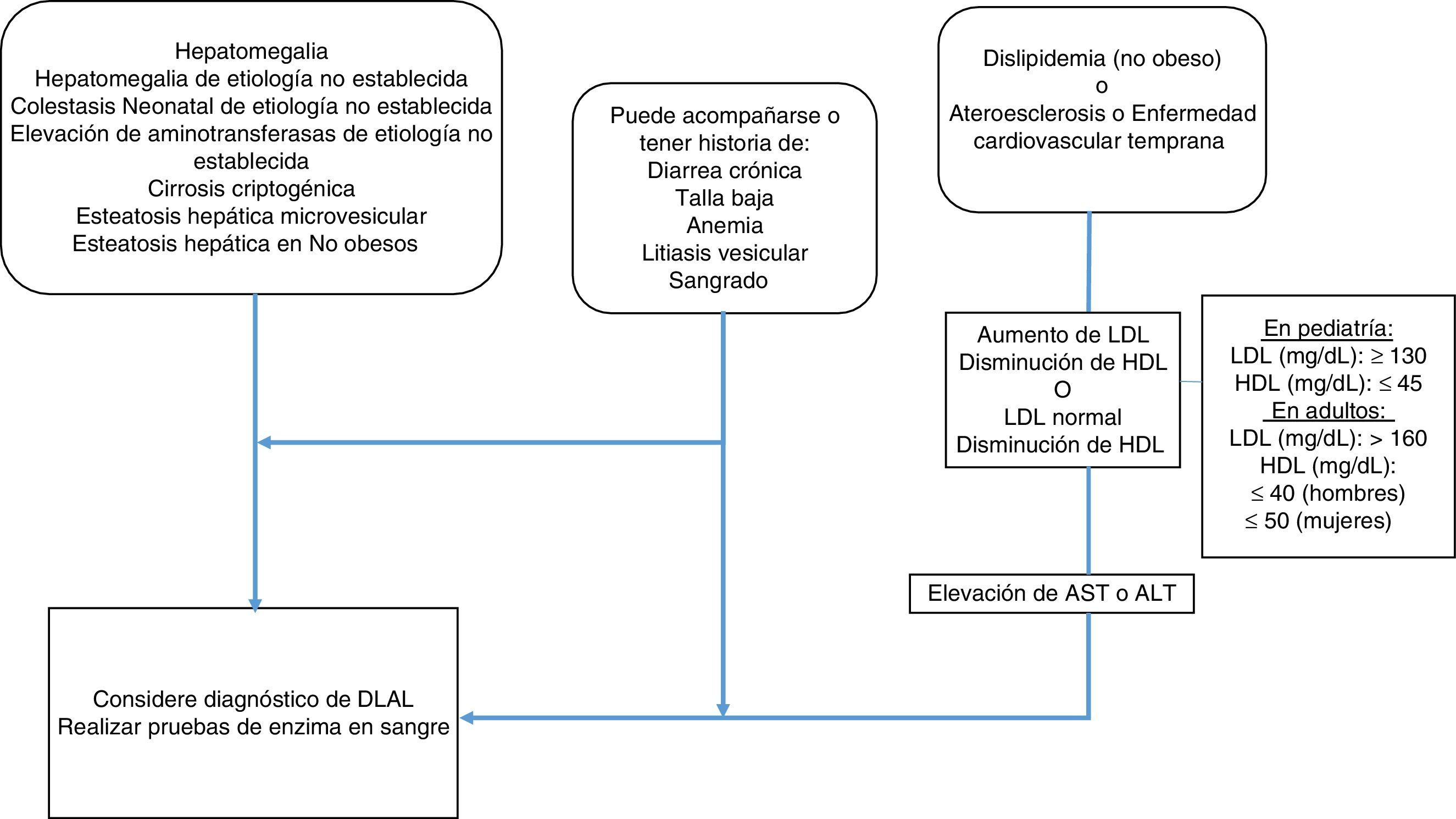
MetodologíaSe realizó una revisión sistematizada de la literatura de las manifestaciones clínicas y el diagnóstico de la DLAL por parte de los expertos mexicanos. Se convocó a 15 especialistas en el área, con conocimiento sobre DLAL para la elaboración de este consenso. Con el objetivo de evaluar lo publicado se realizó una búsqueda de la información a través del siguiente algoritmo de búsqueda en The Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE (PubMed), EMBASE (Ovid), LILACS: («lysosomal acid lipase deficiency»[Mesh] OR «wolman disease» [Mesh] OR «cholesterol ester storage disease» [Mesh]) AND («2000/01/01» [PDAT]: «2016/02/28» [PDAT]) AND «humans» [MeSH Terms] AND (English[lang] OR Spanish[lang]) AND («infant» [MeSH Terms] OR «child» [MeSH Terms] OR «adolescent» [MeSH Terms] OR «adult»[MeSH Terms])). Solamente se evaluaron las publicaciones en idioma español e inglés. Se seleccionaron artículos donde se mencionaran las manifestaciones clínicas, de laboratorio y de patología de la DLAL. Los artículos fueron analizados por los participantes del consenso en 5 grupos: 1) gastroenterólogos (adultos y pediatría), 2) genetistas y Medicina interna, 3) endocrinólogos (adultos y pediatría), 4) Imagenología y 5) Patología; se crearon los enunciados por los grupos; se sometió a votación; aquellos que alcanzaron un valor de consenso de más del 90% fueron incluidos. Con el objetivo de realizar un consenso se utilizó el método cuantitativo estructurado de técnica de grupo nominal para llegar a un consenso. Se procedió a la votación de forma presencial pero anónima, mediante un dispositivo electrónico; aquellos enunciados que superaron el 90% fueron considerados para su inclusión y en base a lo anterior, se estructuró un algoritmo diagnóstico (fig. 1).
Resultados
La búsqueda sistemática realizada mostró 373 artículos, ninguno de los cuales correspondió a una guía de práctica clínica o ensayos clínicos; después de eliminar artículos repetidos, cartas al editor o reportes de casos que ya estaban contemplados en otros artículos quedaron 184 artículos relacionados con manifestaciones clínicas, de laboratorio y de auxiliares de gabinete, los cuales fueron revisados por los participantes.
DefiniciónLa DLAL es una enfermedad por almacenamiento lisosomal, autosómica recesiva ocasionada por mutaciones del gen de la lipasa ácida (LIPA), la disminución de la actividad enzimática se manifiesta como una acumulación progresiva de ésteres de colesterol y triglicéridos en el hígado, bazo y otros órganos1. Se puede presentar como enfermedad hepática progresiva, aterosclerosis temprana o alteraciones gastrointestinales2.
El grado de deficiencia de la enzima lipasa ácida lisosomal (LAL) es variable pero no determina la heterogeneidad del cuadro clínico; hay dos fenotipos: el de presentación temprana, conocido como enfermedad de Wolman, de comportamiento grave, rápidamente progresivo, sin tratamiento los pacientes mueren antes del año de edad y la forma de presentación tardía o enfermedad por depósito de ésteres de colesterol (EDEC), que es menos grave, se manifiesta a cualquier edad, solo el 19% de los casos se presenta en pacientes menores de dos años y el resto en niños mayores y adultos3–6.
Todos los niños con la presentación infantil (enfermedad de Wolman) sin tratamiento, mueren durante el primer año.
EpidemiologíaLa frecuencia de la DLAL en diferentes poblaciones es desconocida debido tal vez al infradiagnóstico7. La incidencia es variable, va desde 1 en 40,000 en Alemania8,9, hasta 1 en 700,000 en otras poblaciones, como Australia2,10. Con base en estas estimaciones, habría 750 a 1,500 pacientes con DLAL tan solo en los EE. UU.10,11.
Los estudios poblacionales que evalúan la presencia de la mutación más frecuente de la EDEC arrojan una frecuencia de portadores de 1 en 1,000 en población asiática y 1 en 300 en poblaciones caucásica e hispana; lo anterior apoya la hipótesis de que la enfermedad está infradiagnosticada.
La DLAL frecuentemente pasa inadvertida.
FisiopatologíaLa LAL es esencial para el metabolismo de los lípidos, hidroliza ésteres de colesterol y triglicéridos de las lipoproteínas de baja densidad (LDL, por sus siglas en inglés), genera colesterol y ácidos grasos libres que a su vez participan en la regulación de la homeostasis del colesterol2,12. En condiciones normales, el exceso de colesterol libre intracelular produce: a) disminución de la actividad de los receptores de LDL, b) inhibición de la actividad enzimática de la 3-hidroxi-3-metilglutaril-coenzima A reductasa, a través de un mecanismo de retroalimentación negativa, lo que disminuye la síntesis de colesterol endógeno y c) estimulación de la acil-colesterol acil-transferasa que incrementa la esterificación de colesterol13.
Al estar disminuida o ausente, la actividad de la enzima LAL, los ésteres de colesterol y triglicéridos se acumulan en los lisosomas; se produce depleción del colesterol libre intracelular, disminución de la capacidad para suprimir la actividad de 3-hidroxi-metilglutaril-coenzima A reductasa y aumento en la producción endógena de colesterol14. Este aumento inhibe la actividad de los receptores de LDL lo que ocasiona disminución en la depuración hepática de las LDL y aumento en la síntesis de apolipoproteína B (ApoB) y de lipoproteínas de muy baja densidad VLDL2. La formación de partículas de lipoproteínas de alta densidad (HDL, por sus siglas en inglés), está mediada por la cantidad de colesterol contenido en los endosomas/lisosomas y por el transportador de colesterol A1 dependiente de la unión de ATP (ABCA1); en estudios realizados en fibroblastos de pacientes con DLAL, se ha encontrado alteración en la regulación de este transportador, que ocasiona disminución en la formación de partículas de HDL14–16.
Estos mecanismos explican la dislipidemia con que cursan los pacientes: elevación de colesterol total (CT) y colesterol unido a las LDL (c-LDL), disminución en las concentraciones de colesterol de las de alta densidad (c-HDL) e hipertrigliceridemia15.
Manifestaciones clínicasEl diagnóstico de esta enfermedad se hace a través de la historia clínica buscando intencionadamente sus síntomas y signos y complementándolo con los análisis de laboratorio correspondientes6,17.
Las manifestaciones clínicas de DLAL de inicio temprano son diversas, destacando la hiporexia, vómitos frecuentes, diarrea, esteatorrea, desnutrición y falla para crecer, y hepatoesplenomegalia2,3.
El daño hepático progresa rápidamente a hipertensión portal y disfunción hepática grave en los primeros meses de vida; los hallazgos de acidosis metabólica y calcificación de las suprarrenales se han descrito en cerca de una tercera parte de los pacientes2,10.
En la DLAL de presentación infantil o enfermedad de Wolman, la disfunción hepática grave es temprana y progresiva.
En niños y adultos, la presentación clínica es variable, la edad promedio de inicio de la sintomatología es de 5 años, pero algunos pacientes se mantienen asintomáticos y se diagnostican hasta la edad adulta. La distensión abdominal que es debida a visceromegalias y la talla baja son comunes1.
Las manifestaciones clínicas en la DLAL de presentación tardía pueden manifestarse con datos inespecíficos como distensión abdominal y talla baja.
Las visceromegalias abdominales son frecuentes en la DLAL cuando se detectan debe considerarse esta enfermedad.
Además de las manifestaciones clínicas de enfermedad hepática crónica y sus complicaciones tales como hipertensión portal, hemorragias, encefalopatía en etapas avanzadas, e incluso carcinoma hepatocelular, se han descrito: dolor abdominal, diarrea con y sin malabsorción, enfermedad vesicular con colelitiasis además de epistaxis de repetición, por trombocitopenia1,15. En la tabla 1 se enlistan los principales hallazgos clínicos de la DLAL de presentación tardía y en la tabla 2 los de laboratorio2,18,19.
Principales manifestaciones clínicas de la DLAL de presentación tardía
| Signos y síntomas clínicos |
| Distensión abdominal |
| Hepatomegalia |
| Hepatoesplenomegalia |
| Falla para crecer |
| Diarrea |
| Esteatorrea |
| Ictericia |
| Enfermedad arterial coronaria |
| Aneurisma |
| Evento vascular cerebral |
| Várices esofágicas |
| Litiasis vesicular |
Principales datos de laboratorio de la DLAL de presentación tardía
| Datos de laboratorio y gabinete |
| Hiperbilirrubinemia |
| Elevación de enzimas hepáticas |
| Prolongación del tiempo de protrombina |
| Niveles elevados de colesterol y de triglicéridos |
| Niveles elevados de lipoproteínas de baja densidad |
| Niveles disminuidos de lipoproteínas de alta densidad |
| Anemia |
| Trombocitopenia |
| Hígado graso identificado por algún método imagenológico |
| Esteatosis hepática microvesicular |
Se debe descartar DLAL de presentación tardía en pacientes con hepatoesplenomegalia, elevación persistente de las aminotransferasas y dislipidemia.
La elevación de aminotransferasas por hallazgo accidental obliga a buscar crecimiento de hígado y bazo.
DislipidemiaLas principales manifestaciones cardiovasculares son: enfermedad arterial coronaria, aneurisma y accidente vascular cerebral1,20. Las complicaciones relacionadas con enfermedad cardiovascular aterosclerótica de inicio temprano están asociadas a la dislipidemia secundaria a alteraciones hepáticas del metabolismo de los lípidos21. En diversos estudios se ha encontrado que las complicaciones asociadas a enfermedad cardiovascular aterosclerótica de inicio temprano se presentan en 4.2% de los pacientes con DLAL y 12.5% de pacientes mayores de 25 años con DLAL15,22,23.
La dislipidemia asociada a DLAL de presentación tardía está relacionada con aterosclerosis y es un factor de riesgo para enfermedad cardiovascular temprana.
La dislipidemia en niños y adultos con DLAL de presentación tardía se caracteriza por concentraciones elevadas de colesterol total, LDL, y ApoB y disminución del HDL. Algunos casos pueden cursar con hipertrigliceridemia.
Estudios de laboratorioBioquímicamente, la mayoría de los pacientes con DLAL cursan con elevación de alaninoaminotransferasa y aspartatoaminotransferasa, hiperbilirrubinemia leve, gammaglutamiltransferasa elevada, tiempo de protrombina prolongado, hipoglucemia en ayunos prolongados y dislipidemia IIA o IIB, con hipercolesterolemia, disminución del c-HDL, elevación del c-LDL, ApoB y en ocasiones hipertrigliceridemia24. Las concentraciones de LDL generalmente son más elevadas que en los pacientes con enfermedad hepática por hígado graso no alcohólico en concentraciones promedio de 100-200mg/dl; esta elevación no es tan alta como en los pacientes con hipercolesterolemia familiar heterocigota. Se pueden identificar datos de anemia en 12.5% de los casos, así como trombocitopenia y leucopenia, si cursa con hepatoesplenomegalia o cirrosis hepática. La elevación de la alaninoaminotransferasa se ha observado en 91.7% de los pacientes, la de aspartatoaminotransferasa en 76.2%, de gammaglutamiltransferasa en un 20% y elevación de la bilirrubina total en 20%15.
En todo paciente con enfermedad hepática se recomienda realizar un perfil de lípidos completo además del monitoreo periódico de las aminotransferasas por lo menos durante seis meses.
ImagenologíaEn la presentación temprana, el 50% de los niños presentan calcificación de las glándulas suprarrenales, en una radiografía simple1,2.
La calcificación de las glándulas suprarrenales es característica de DLAL de presentación infantil.
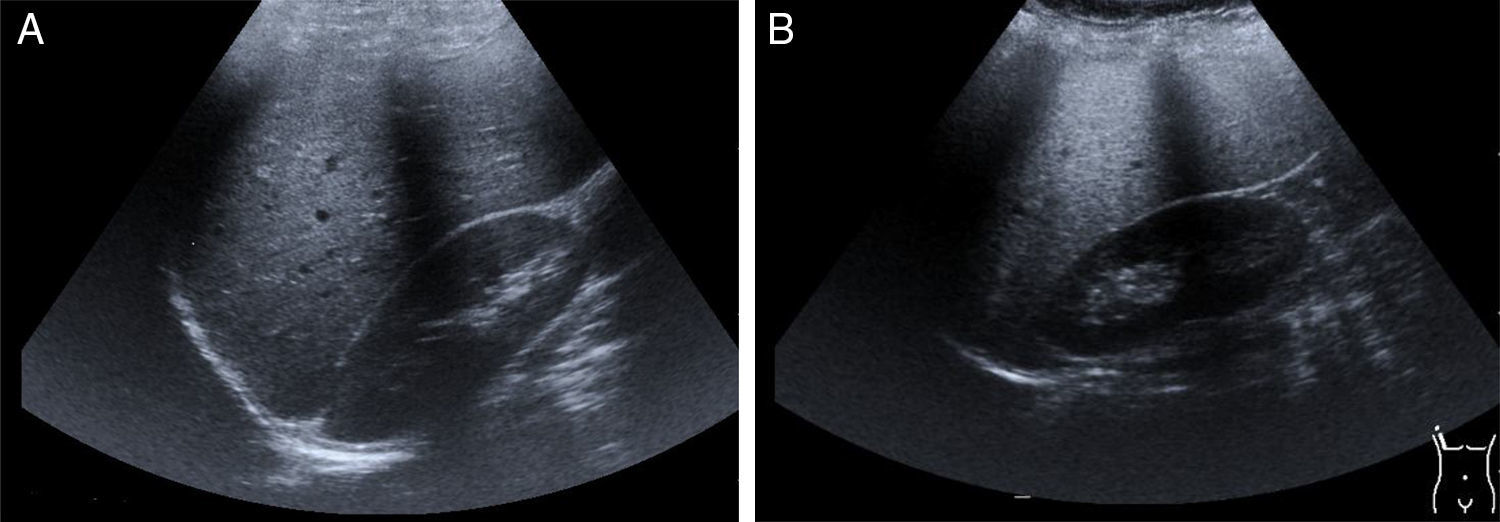
Los hallazgos de los estudios de imagen corroboran la hepatomegalia, esplenomegalia, e identifica datos de esteatosis hepática. Los estudios de ultrasonido, tomografía axial computarizada, resonancia magnética con espectroscopia, la elastografía de transición o la fusión de la elastografía y resonancia magnética, son herramientas diagnósticas25 no invasivas utilizadas para la evaluación y estadificación del hígado graso (fig. 2 A y B).
 Imagen de hígado normal obtenida por ultrasonido. B) Se observa hígado con esteatosis. Es posible identificar hepatomegalia.")
La DLAL de presentación tardía es una causa de esteatosis hepática identificable por ultrasonido.
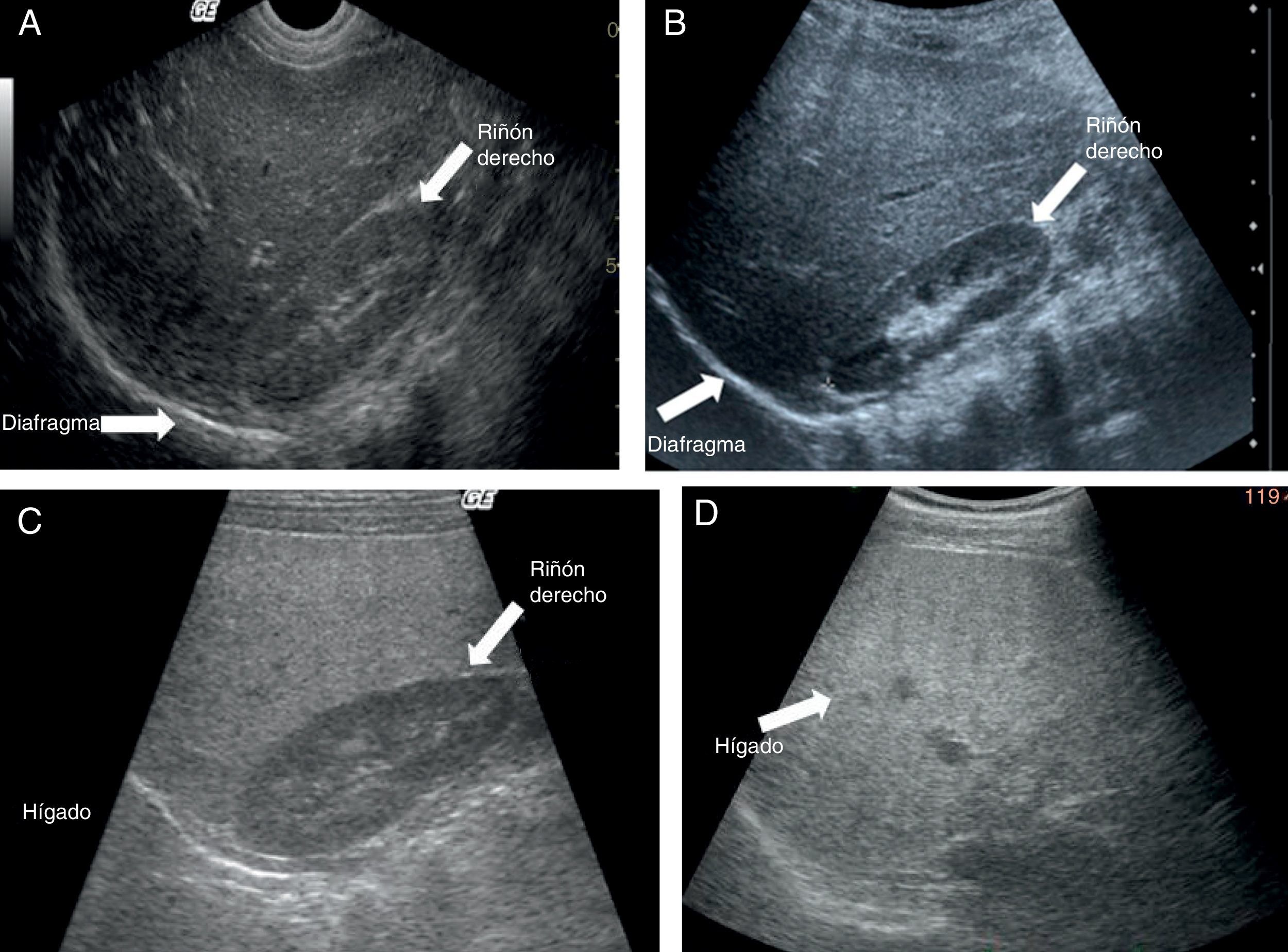
El ultrasonido muestra ecogenicidad difusa, hepatomegalia y esplenomegalia (fig. 3A). La clasificación de Tominaga26 para evaluar la ecogenicidad hepática comprende tres estadios: a) leve, con mínimo aumento difuso de la ecogenicidad hepática, aspecto normal del diafragma y de los vasos intrahepáticos (fig. 3B), b) moderada, aumento discreto y difuso de la ecogenicidad hepática; ligera disminución de los vasos intrahepáticos y del diafragma (fig. 3C) y c) grave, aumento de la ecogenicidad, pobre penetración del segmento posterior del lóbulo hepático derecho y pobre o nula identificación de los vasos hepáticos y del diafragma (fig. 3D).
 Imagen de ultrasonido que demuestra ecogenicidad conservada en hígado y riñón derecho. B) Imagen de ultrasonido que demuestra discreto incremento de la ecogenicidad hepática (esteatohepatitis leve). C) Imagen de ultrasonido que demuestra importante incremento en la brillantez hepática (esteatohepatitis moderada). D) Imagen de ultrasonido que demuestra hígado ecogénico. No se identifican vasos porta (esteatohepatitis grave).")
A) Imagen de ultrasonido que demuestra ecogenicidad conservada en hígado y riñón derecho. B) Imagen de ultrasonido que demuestra discreto incremento de la ecogenicidad hepática (esteatohepatitis leve). C) Imagen de ultrasonido que demuestra importante incremento en la brillantez hepática (esteatohepatitis moderada). D) Imagen de ultrasonido que demuestra hígado ecogénico. No se identifican vasos porta (esteatohepatitis grave).
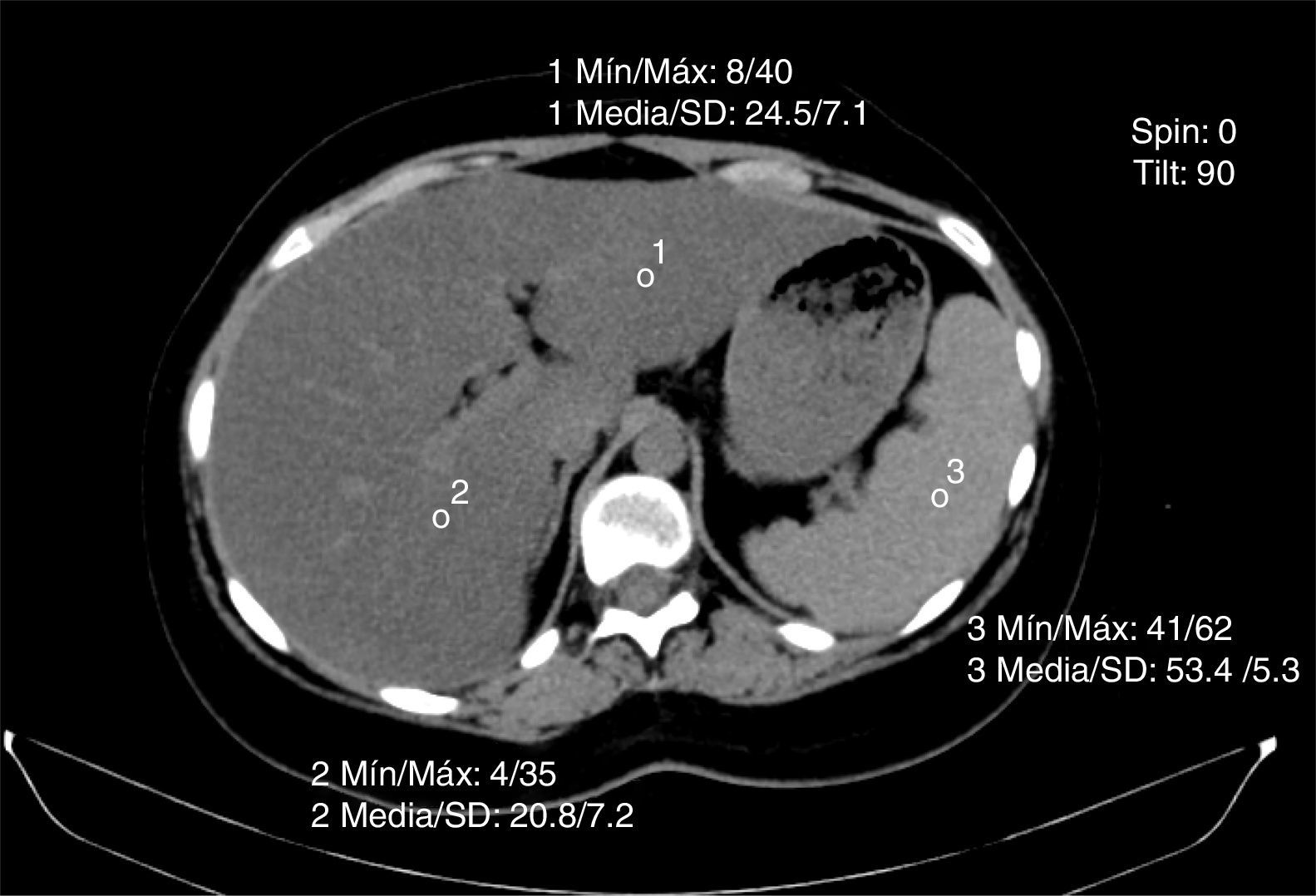
La tomografía axial computarizada en fase simple valora la atenuación hepática medida con unidades Hounsfield, valores de atenuación entre hígado-bazo27. Si estos valores se sitúan por debajo de 48 unidades Hounsfield se diagnostica infiltración grasa (fig. 4). La optimización de la resonancia magnética y la elastografía son también métodos diagnósticos al igual que la espectroscopia de resonancia magnética nuclear.

La determinación de isótopos de hidrógeno (1H) y de carbono (13C) en tejido graso difiere en pacientes con DLAL en comparación con pacientes con EHGNA, debido a la relación molar dada por la cantidad de grupos metilo (CH3) en proporción al grupo metileno (CH2); una fracción de colesterol tiene cinco grupos CH3 con una fuerte resonancia en el espectro de resonancia magnético de 1H, comparado con una fracción de ácido graso que tiene un grupo metil por molécula28. En pacientes con DLAL, la concentración media de colesterol en tejido hepático fue de 21.1±13.6mM, y de ácidos grasos de 51.2±19.2mM; mientras que en pacientes con EHGNA, fue de 442±164mM para ácidos grasos28. Esto traduce una correlación entre la determinación bioquímica y la determinación de colesterol y ácidos grasos por medio de este método el cual puede ser útil como alternativa, no invasiva, para determinar colesterol y triglicéridos hepáticos e, incluso, como prueba de vigilancia de la enfermedad.
PatologíaEl estudio histopatológico de las biopsias de los pacientes con DLAL ofrece, al menos, tres beneficios: a) por sí solo puede ser indicativa de la enfermedad, b) contribuye a confirmar el diagnóstico cuando se sospecha esta entidad y c) proporciona información valiosa para establecer el pronóstico; además, es de utilidad para el seguimiento de los pacientes, la evaluación del tratamiento y para el análisis retrospectivo29.
Las alteraciones histopatológicas de la DLAL son características y varían en su intensidad de acuerdo con la edad, tiempo de evolución y expresividad de la enfermedad; los órganos principalmente afectados son el hígado y todos aquellos en donde la presencia de macrófagos es predominante1,7. La presentación infantil, tiene manifestaciones floridas no solo en el hígado, sino en los diversos órganos que se encuentran extensamente afectados1.
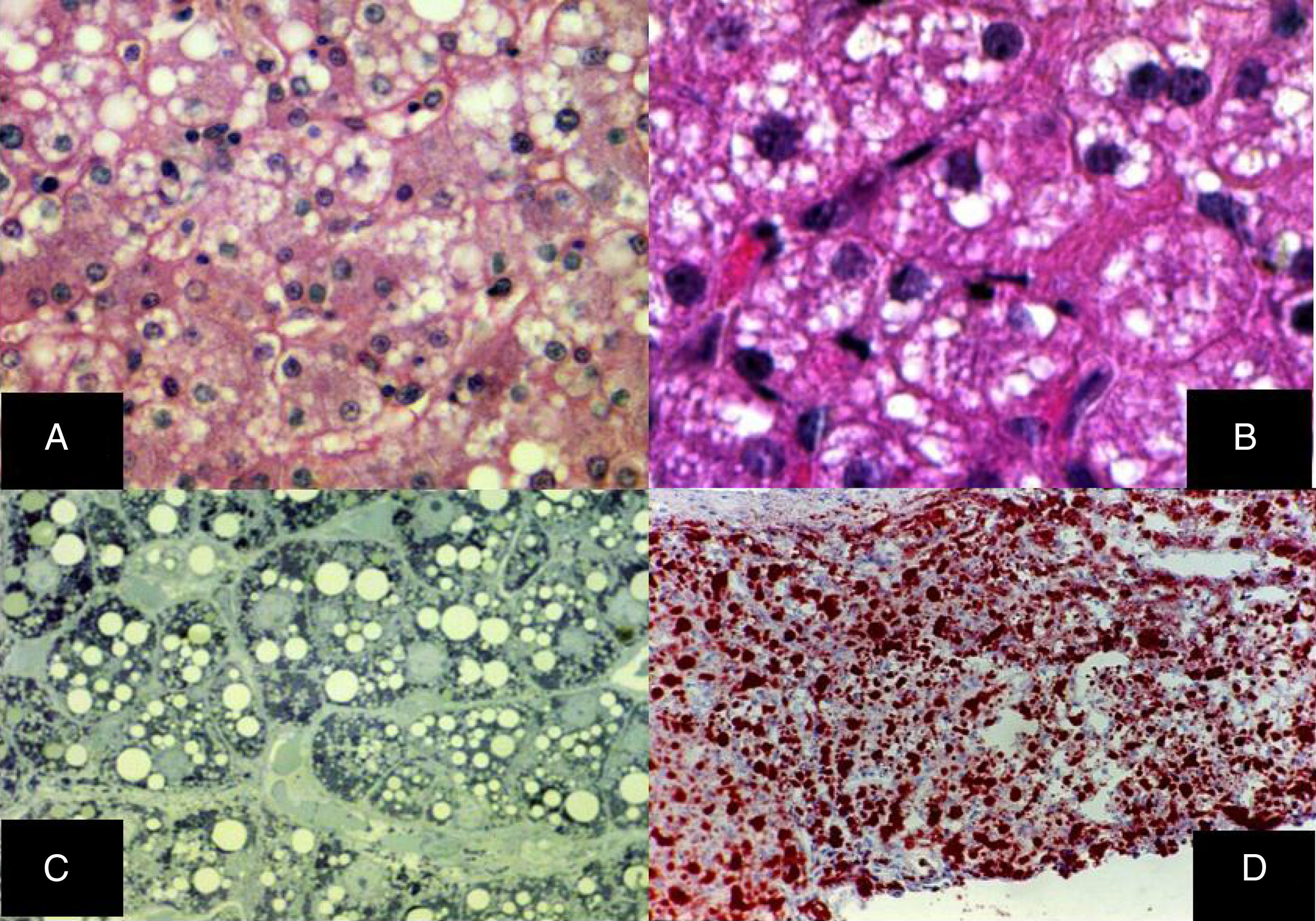
En el hígado, la lesión fundamental es la esteatosis microvesicular difusa, el citoplasma de los hepatocitos contiene numerosas gotas de lípidos intensamente positivas con la tinción de rojo oleoso; estas vesículas son de menos de una micra de diámetro y no desplazan el núcleo (fig. 5)29–32.
 Esteatosis microvesicular difusa, se aprecian múltiples vacuolas claras dentro del citoplasma de los hepatocitos (H&E, aumento original x 400). B) Las vacuolas miden alrededor de una micra de diámetro y no desplazan al núcleo (H&E, aumento original x 400). C) Cortes semifinos que muestran la naturaleza microvesicular de la esteatosis, la cual se aprecia en prácticamente todos los hepatocitos (azul de toluidina, aumento original x 400). D) Múltiples gotas de grasas neutras se revelan en todo el lobulillo hepático (rojo oleoso, aumento original x 200).")
A) Esteatosis microvesicular difusa, se aprecian múltiples vacuolas claras dentro del citoplasma de los hepatocitos (H&E, aumento original x 400). B) Las vacuolas miden alrededor de una micra de diámetro y no desplazan al núcleo (H&E, aumento original x 400). C) Cortes semifinos que muestran la naturaleza microvesicular de la esteatosis, la cual se aprecia en prácticamente todos los hepatocitos (azul de toluidina, aumento original x 400). D) Múltiples gotas de grasas neutras se revelan en todo el lobulillo hepático (rojo oleoso, aumento original x 200).
Los hallazgos en la biopsia hepática sugestivos de DLAL de presentación tardía son esteatosis microvesicular difusa y cristales de colesterol en el citoplasma de los hepatocitos y macrófagos.
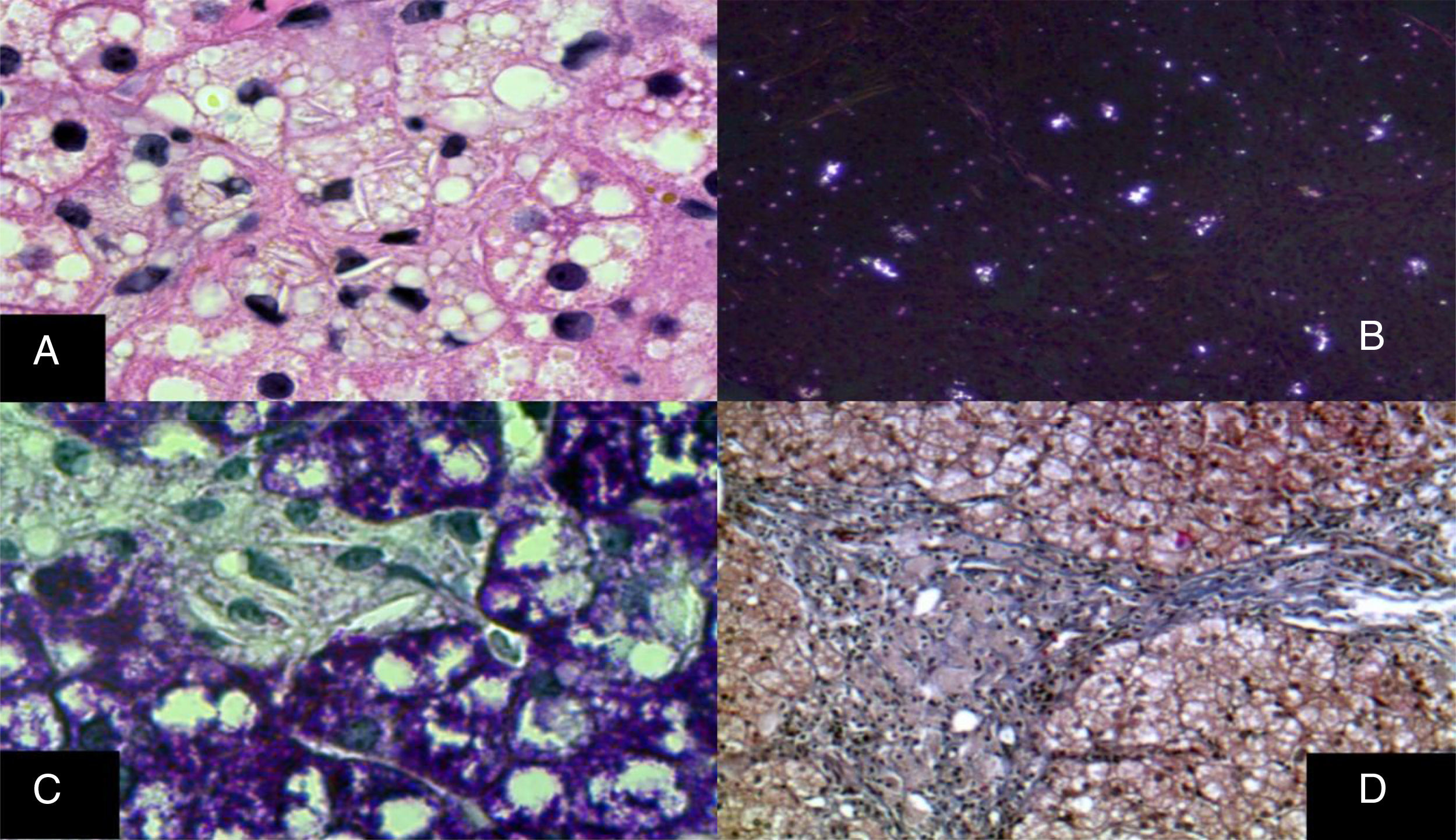
La lesión del sistema monocito macrófago afecta a las células de Kupffer y a los macrófagos portales; ambas muestran pequeñas gotas en su citoplasma y cristales de colesterol; además, los macrófagos acumulan pigmento parecido a la lipofuscina (fig. 6)29–33. Los hepatocitos pueden tener en su citoplasma cristales de colesterol que exhiben una refringencia plateada característica cuando se observan bajo luz polarizada29–33.
 Reacción de inmunohistoquímica en donde se observa que in situ tanto en los hepatocitos como en las células de Kupffer se aprecian hendiduras claras en su citoplasma que corresponden a cristales de colesterol (H&E, aumento original x 1,000). B) Birefrigerancia plateada característica de los numerosos cristales de colesterol (luz polarizada, aumento original x 100). C) Las células de Kupffer con esteatosis microvesicular y cristales de colesterol destacan claramente (PAS, aumento original x 1,000). D) Se muestra un espacio porta expandido por numerosos macrófagos de citoplasma de aspecto «espumoso» dado por las inclusiones de lípidos (tricrómico de Masson, aumento original x 200).")
A) Reacción de inmunohistoquímica en donde se observa que in situ tanto en los hepatocitos como en las células de Kupffer se aprecian hendiduras claras en su citoplasma que corresponden a cristales de colesterol (H&E, aumento original x 1,000). B) Birefrigerancia plateada característica de los numerosos cristales de colesterol (luz polarizada, aumento original x 100). C) Las células de Kupffer con esteatosis microvesicular y cristales de colesterol destacan claramente (PAS, aumento original x 1,000). D) Se muestra un espacio porta expandido por numerosos macrófagos de citoplasma de aspecto «espumoso» dado por las inclusiones de lípidos (tricrómico de Masson, aumento original x 200).
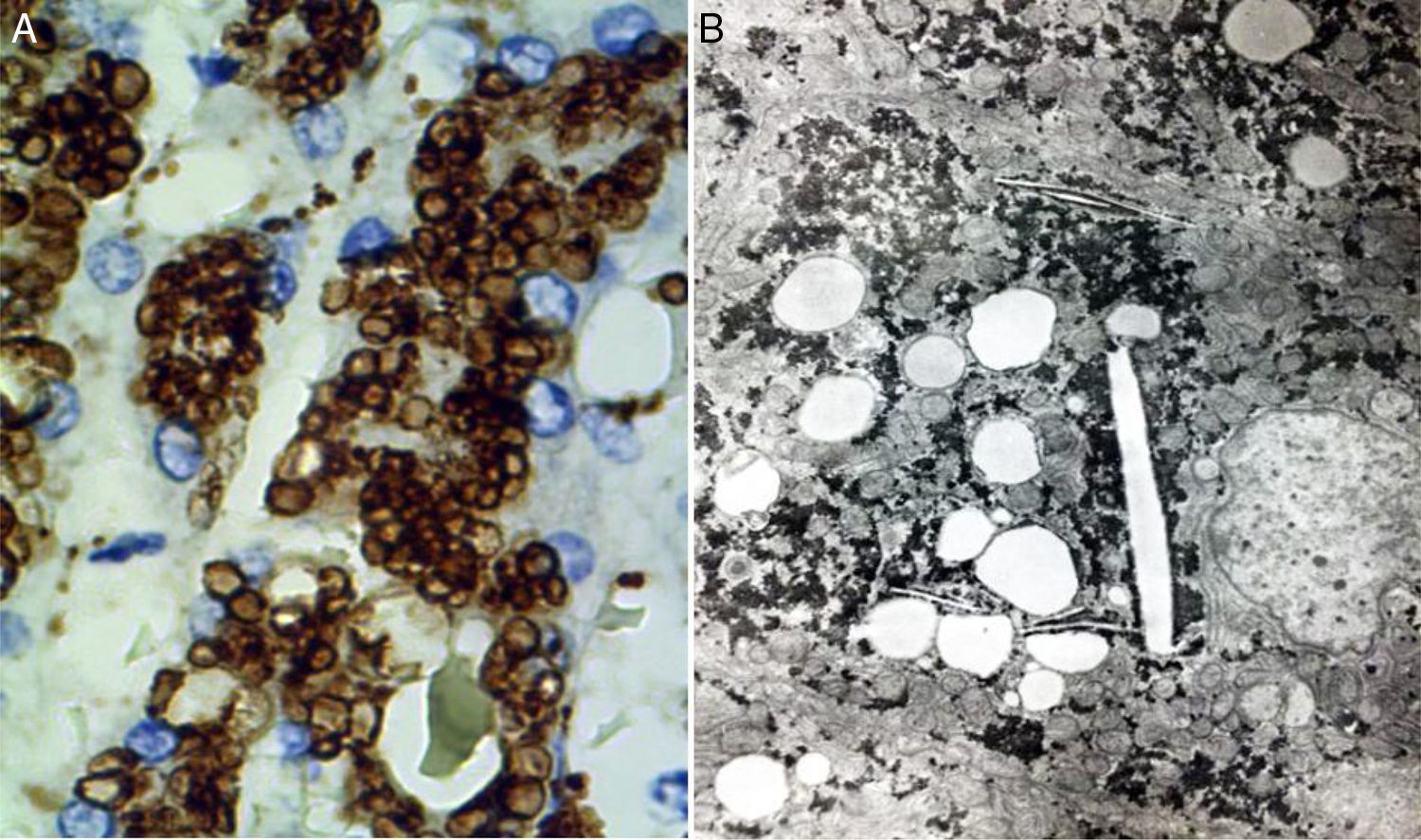
Para demostrar que los lípidos se encuentran dentro de lisosomas, se cuenta con dos herramientas fundamentales: la inmunohistoquímica y la microscopia electrónica. Se pueden utilizar anticuerpos que reconocen a componentes lisosomales tales como la proteína integral de membrana lisosomal 2 (LIMP2), proteína asociada a la membrana lisosomal 1 (LAMP1) y la proteína luminal lisosomal o catepsina D (fig. 7A).
 En esta reacción de inmunohistoquímica se observa que las gotas de lípidos se encuentran en lisosomas (catepsina D, aumento original x 400). B) En esta fotomicrografía se aprecia que los lípidos están rodeados de una unidad simple de membrana que revela su naturaleza lisosomal; se aprecian también cristales de colesterol (microscopía electrónica, aumento original x 2,000).")
A) En esta reacción de inmunohistoquímica se observa que las gotas de lípidos se encuentran en lisosomas (catepsina D, aumento original x 400). B) En esta fotomicrografía se aprecia que los lípidos están rodeados de una unidad simple de membrana que revela su naturaleza lisosomal; se aprecian también cristales de colesterol (microscopía electrónica, aumento original x 2,000).
Bajo el microscopio electrónico se observa que los lípidos están en el interior de los lisosomas (fig. 7B) rodeados de una unidad simple de membrana y no acumuladas libremente en el citosol. La fibrosis hepática es dependiente del tiempo de evolución y de la expresividad del padecimiento; esta puede variar desde leve a cirrosis29,31.
En los pacientes con esteatosis predominantemente microvesicular demostrada por biopsia se debe descartar DLAL de presentación tardía mediante inmunohistoquímica o microscopía electrónica.
Este cortejo histopatológico y la demostración del almacenamiento de lípidos en los lisosomas, permite hacer el diagnóstico, y diferenciar otras entidades que cursan con esteatosis microvesicular entre ellas otras enfermedades por almacenamiento29,30, como se señala en el tabla 2.
En los pacientes adultos con cirrosis hepática criptogénica no obesos se debe de descartar DLAL de presentación tardía.
Diagnóstico enzimático y molecular de DLALLa LAL (E.C.3.1.1.13) también conocida como colesterol éster sintasa, colesterol esterasa, triterpenol esterasa, lipasa A y esteril éster acil hidrolasa, es la enzima encargada de hidrolizar los ésteres de colesterol y los triglicéridos de las lipoproteínas de baja densidad, generando colesterol y ácidos grasos libres2; por lo tanto, su deficiencia lleva a la acumulación de lípidos y a las alteraciones en la homeostasis lipídica34,35.
El diagnóstico bioquímico de la DLAL en leucocitos, fibroblastos y líquido amniótico se conoce desde hace 50 años36,37 y en 2012, se incorporó el uso de un inhibidor (Lalistat 2) que mejoró la especificidad de la técnica38–40.
La prueba en gota de sangre seca en papel filtro (GSSPF), se puede utilizar en pacientes de cualquier edad, permite determinar la actividad de LAL y es útil para detectar pacientes afectados2, su sensibilidad y especificidad es muy alta38. Para realizarla, se obtiene una pequeña cantidad de sangre por punción; se coloca inmediatamente en el papel filtro Whatman 903 que la absorbe y se deja secar hasta el día siguiente a temperatura ambiente, se recomienda no exponer la muestra al calor y transportarla en bolsas dobles con desecante para mantener la actividad de la enzima; se puede almacenar a largo plazo a - 20°C41,42. Los valores normales de la actividad de la LAL dependen del laboratorio donde se haga la determinación. Se realizó un consenso de expertos a nivel mundial para el diagnóstico de LAL, en el que se determinó la importancia de analizar los resultados de la actividad de LAL en base al porcentaje de actividad con respecto a la media normal de la población sana de referencia analizada por cada laboratorio, lo cual permite identificar a los pacientes afectados, quienes presentan un valor de actividad enzimática menor al 5%, aquellos que se encuentran en zona gris cuando los valores son entre el 5% y el 10% de la actividad enzimática y los no afectados con una actividad enzimática >10% respecto de la media de la población sana43. Las ventajas son: pequeña cantidad de sangre, facilidad para la toma de muestra, estabilidad adecuada en condiciones ambientales; además de que favorece el diagnóstico en poblaciones grandes en forma rápida, se maneja fácilmente y el costo es bajo37,38,41,42.
En México se lleva a cabo en el Laboratorio de Gastroenterología en el Hospital Infantil de México Federico Gómez, y en el Centro de Investigación Biomédica de Occidente, en Guadalajara.
El diagnóstico enzimático de DLAL puede hacerse en una gota de sangre seca en papel filtro.
El diagnóstico molecular de DLAL requiere del análisis de la secuencia del gen LIPA, localizado en 10q23.31, el cual contiene 10 exones, mide 36kb y produce una proteína de 399 aminoácidos; se hace en leucocitos de sangre periférica e incluye la secuenciación del gen o de un panel de genes que lo incluyen. El ADN se extrae de la muestra (leucocitos o GSSPF) para la secuenciación del gen o secuenciación masiva9,40,44,45.
Cuando el diagnóstico por secuenciación o panel de genes no confirma el diagnóstico se debe considerar el abordaje genómico40; a la fecha se han detectado más de 40 mutaciones en el gen LIPA1,40,46–49; la más frecuente (50-70%), se localiza en el sitio de corte y unión del exón 8 (E8SJM1G>A); en México, se han encontrado las mutaciones c.253C>A y c.294C>G en pacientes con fenotipo EDEC. Se debe considerar al diagnóstico molecular como de soporte, ya que en algunos casos no se puede identificar la o las mutaciones responsables; además, el costo es elevado. La determinación de la actividad enzimática en papel es una prueba accesible, precisa y de menor costo.
Diagnóstico diferencialDada la variabilidad de la DLAL se debe diferenciar en la etapa neonatal de las diversas causas de colestasis neonatal, de las cuales destacan claramente las enfermedades metabólicas, principalmente aquellas que cursan con insuficiencia hepática de rápida evolución, como tirosinemia, galactosemia neonatal, hemocromatosis y la enfermedad de Niemann-Pick C50–53.
Se debe considerar a la DLAL en el diagnóstico diferencial de la colestasis neonatal con insuficiencia hepática.
Por el hecho de que la hiperlipidemia observada en pacientes con DLAL es del tipo IIA o IIB, en el diagnóstico diferencial de esta enfermedad se debe considerar hipercolesterolemia familiar heterocigota, ApoB100 familiar defectuosa, hiperlipidemia familiar combinada e hipercolesterolemia poligénica11,54,55.
La hipercolesterolemia familiar se puede confundir con la DLAL dado que en las dos se encuentra LDL elevado; sin embargo, en la hipercolesterolemia familiar, no se observa hepatoesplenomegalia. La hipercolesterolemia familiar se hereda en forma autosómica co-dominante mientras que la DLAL se hereda en forma AR56,57.
En todo niño o adulto con sospecha de hipercolesterolemia familiar heterocigota, debe considerarse como diagnóstico diferencial la DLAL de presentación tardía.
Debido a que los pacientes con DLAL presentan hepatomegalia, elevación de enzimas hepáticas, hígado graso y dislipidemia, se debe considerar el síndrome metabólico como diagnóstico diferencial, que presenta un mayor riesgo de enfermedad cerebrovascular y diabetes mellitus tipo 258. Solo 27.1% de los pacientes con DLAL presenta hipertrigliceridemia, pero no cursan con obesidad2,15.
En individuos no obesos con o sin síndrome metabólico con hepatomegalia, elevación de aminotransferasas, hígado graso y dislipidemia, se debe pensar en DLAL de presentación tardía.
ConclusiónLa DLAL es una enfermedad poco conocida y por tanto no se piensa en ella, lo que puede condicionar que pase desapercibida o se establezca un diagnóstico erróneo al confundirse con otras enfermedades. El algoritmo propuesto proporciona una secuencia de acciones, basado en las manifestaciones clínicas recopiladas de los diferentes estudios y tiene el objetivo de optimizar el proceso diagnóstico de los pacientes con DLAL.
AlgoritmoDado el fenotipo heterogéneo de la DLAL, no es necesario que un paciente cumpla con todos los criterios enumerados previamente para tener en cuenta a esta enfermedad en el diagnóstico diferencial. Los criterios de sospecha de DLAL están planteados considerando dos grandes grupos; aquel que se presenta con cualquier entidad que genera afección hepática y aquel que se manifiesta con un cuadro de dislipidemia/aterosclerosis/enfermedad cardiovascular temprana. Ambos grupos pueden acompañarse o tener historia de diarrea crónica, talla baja, anemia, litiasis vesicular o hemorragia.
En el grupo de pacientes con afección hepática sin etiología clara se recomienda considerar el diagnóstico de DLAL y realizar la prueba de papel filtro para la determinación de la LAL en sangre.
En aquel grupo donde la presentación es principalmente dislipidemia/aterosclerosis/ enfermedad cardiovascular temprana, se recomienda identificar si la dislipidemia es de tipo II A/B; en donde existe aumento de c-LDL y disminución de c-HDL siempre se deben considerar valores de referencia para la población pediátrica, no obstante hay que tener en cuenta que también podría encontrarse c-LDL normal y c-HDL por debajo de valores normales, sobre todo en pacientes con enfermedad hepática, por lo que se recomienda determinar las aminotransferasas y si están elevadas debe realizarse la determinación de la LAL en papel filtro en sangre.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo/estudio.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

