
El síndrome de Peutz Jeghers (SPJ) es una enfermedad rara con herencia autosómica dominante, causada por una mutación germinal del gen STK11/LKB1, localizado en el cromosoma 19p13.3, que consiste en hiperpigmentación mucocutánea, poliposis hamartomatosa y predisposición al cáncer. Se pretende identificar y registrar a los pacientes con SPJ, hacer una descripción de la enfermedad y estimar la prevalencia en Valencia (España).
Materiales y métodosSe obtuvo un listado de historias clínicas en 10 hospitales, utilizando el conjunto mínimo de base de datos de ingresos hospitalarios del Ministerio de Sanidad y Consumo de España utilizando el código CIE-9 759.6.
ResultadosSe encontraron 405 historias clínicas, de las que 15 pacientes estaban afectados (9 hombres y 6 mujeres), edad media de diagnóstico y fallecimiento de 13.8 y 54.2 años respectivamente, fallecieron 4 hombres, todos por cáncer; la prevalencia estimada es de 0.4/100,000 habitantes. Todos tenían anemia y pólipos en el intestino delgado (80% duodeno, 66.7% íleon y 40% en el yeyuno), el 93.3% tuvieron una intervención quirúrgica urgente y una invaginación intestinal, el 40% de pacientes desarrollaron cáncer con una edad media de 48.5años.
ConclusiónEste es el primer estudio de registro de pacientes con SPJ en Valencia. La codificación CIE-9 es inespecífica para una enfermedad rara. La localización más frecuente de los pólipos fue el duodeno. La mayoría de casos ha tenido una invaginación intestinal, obstrucción intestinal e intervenciones quirúrgicas urgentes; un porcentaje elevado de pacientes presentó cáncer. Interesaría realizar un seguimiento y evaluación de los protocolos existentes en la Comunidad Valenciana.
Peutz-Jeghers syndrome is a rare autosomal dominant inherited disease caused by a germline mutation of the STK11/LKB1 gene, located on chromosome 19p13.3. It is characterized by mucocutaneous hyperpigmentation, hamartomatous polyposis, and predisposition to cancer. The aim of the present study was to identify and register patients with Peutz-Jeghers syndrome, describe the disease, and estimate its prevalence in Valencia (Spain).
Materials and methodsA print-out of the clinical histories from 10 hospitals was obtained utilizing the ICD-9 code 759.6 from the Minimum Basic Data Set of Hospital Admissions of the Spanish Ministry of Health and Consumer Affairs.
ResultsFrom a total of 405 clinical histories found, 15 (9 males and 6 females) fit the diagnostic criteria of Peutz-Jeghers syndrome. Mean age at diagnosis was 13.8 years and mean age at death was 54.2 years. Four males died, all from cancer. The estimated disease prevalence was 0.4/100,000 inhabitants. All the patients presented with anemia and polyps in the small bowel (80% in the duodenum, 66.7% in the ileum, and 40% in the jejunum), 93.3% underwent urgent surgical intervention and presented with intestinal invagination, and 40% of the patients developed cancer at a mean age of 48.5 years.
ConclusionThe present study is the first register of patients with Peutz-Jeghers syndrome in Valencia, Spain. The ICD-9 code is nonspecific for rare diseases. The duodenum was the most frequent location for polyps and the majority of cases presented with intestinal invagination, bowel obstruction, and urgent surgical intervention. A large percentage of patients presented with cancer. It would be of interest to review and evaluate the existing surveillance protocols in the Valencian Community.
El síndrome de Peutz Jeghers (SPJ) se caracteriza por la presencia de poliposis hamartomatosa del tracto gastrointestinal, hiperpigmentación mucocutánea y predisposición a presentar cáncer; es una enfermedad genética que se encuentra dentro del grupo de las enfermedades raras (ER), estas enfermedades son definidas en la Unión Europea como aquellas cuya cifra de prevalencia se encuentra por debajo de 5 casos por cada 10,000 personas, además tienen una alta tasa de mortalidad y baja prevalencia1,2. Presenta un pico de incidencia entre los 10 y los 30 años3,4. Históricamente el médico Connor JT habló por primera vez de la descripción de este síndrome en el año 1895 en unas gemelas idénticas de 12 años de edad que presentaban pigmentación en los labios y en la boca; una murió a los 20 años por obstrucción intestinal y la segunda a los 52 años por cáncer de mama5. El médico holandés Peutz (1921) describió a 7 miembros de una familia holandesa (familia Harrisburg), que presentaban pigmentación mucocutánea y poliposis en el intestino y la nasofaringe, con antecedentes de 2 muertes en 2 mujeres antes de los 20 años secundarias a obstrucción intestinal6. En 1924 Van Dijk y Oudenal describieron 2 hermanos con pigmentación mucocutánea característica en la boca, poliposis intestinal, obstrucción intestinal y múltiples cirugías abdominales7,8. Posteriormente Jeghers et al. (1949) describieron 10 casos de diferentes familias y publicaron las características del síndrome9. El epónimo de «otros diagnósticos» fue otorgado por Bruwer et al., de la Clínica Mayo, en el año 19545,10. Esta enfermedad tiene una amplia heterogeneidad genética y penetrancia variable e incompleta. La mutación causante se encuentra en línea germinal del gen supresor de tumores STK11/LKB1, localizado en la región telomérica del brazo corto del cromosoma 19p13.311, el cual codifica para una proteína serina/treonina quinasa (liver kinasa B1 [Lkb1]); esta tiene varios papeles dependiendo la vía molecular en que se implique. Aún no está definida la relación entre genotipo y fenotipo, se concluye que existe una amplia heterogeneidad genética12–17. Se ha confirmado que la presencia de una mutación causa pérdida de expresión o función de STK11, una proteína acortada o una proteína truncada con dominios catalíticos incompletos, dando como resultado la pérdida de actividad catalítica quinasa y de su funcionalidad18,19. El papel de STK11 es actuar como un regulador negativo (inhibidor) en varias vías metabólicas, y su alteración conlleva la subsecuente disminución en la inhibición del crecimiento y desarrollo celular, obteniendo como resultado un crecimiento incontrolado de las células y explicando de esta forma el desarrollo de poliposis intestinal y tumores. Las alteraciones secundarias entre la interacción entre Lkb1 y AMPK engloban diferentes procesos que finalmente producen efectos en la función de STK11 sobre la supresión del crecimiento celular y la regulación de la homeostasis energética de la célula.
Sus manifestaciones clínicas incluyen lesiones mucocutáneas hiperpigmentadas (85-99%)12,13,20, poliposis generalmente gastrointestinal de tipo hamartomatoso que asocia la presentación de síntomas y complicaciones como dolor abdominal, sangrados gastrointestinales, anemia secundaria, prolapso y ulceración de un pólipo, obstrucción intestinal e invaginación intestinal; estas 2 últimas la mayoría de ocasiones requieren de laparotomías de urgencia y resecciones intestinales por isquemia14,21. Además estos pacientes asocian un riesgo elevado de padecer cáncer gastrointestinal y extraintestinal. Las lesiones mucocutáneas varían en su forma de presentación; pueden estar presentes desde el nacimiento o aparecen en la infancia, pueden desaparecer en la pubertad, están localizadas más frecuentemente en la mucosa oral (66-83%), ya sea en el paladar o en las encías (pero no en la lengua)5, los labios y la zona perioral (94-96%), alrededor de los ojos y la nariz (36%), a nivel periocular en donde la pigmentación se observa en los párpados y sus bordes y a veces en la conjuntiva palpebral; además en los dedos, la cara dorsal y volar de las manos (74%) y los pies (62%), algunas ocasiones en la región perianal, genital y la mucosa intestinal. Estas son típicamente planas, pueden aparecer como pecas esparcidas o como manchas solitarias de color gris-azulado o marrones, y aproximadamente miden de 1 a 5mm de diámetro, y las lesiones raramente se malignizan5,10,16,17,20,22,23.
La siguiente manifestación más frecuente es la presentación de pólipos intestinales. Algunos pacientes solo manifiestan hiperpigmentación, mientras que otros manifiestan tanto hiperpigmentación como pólipos intestinales24. Son encontrados en todo el tracto gastrointestinal, pero con más frecuencia en el intestino delgado (ID) (60-90%), en orden de frecuencia se encuentran en el yeyuno, el íleon y el duodeno, seguidos por el colon (50-63%), estómago (49%) y recto (32%)17, además pueden aparecer en otras localizaciones anatómicas; generalmente se desarrollan alrededor de la adolescencia temprana y son la causa de gran parte de la morbilidad clínica25,26.
Histológicamente los pólipos del SPJ son de tipo hamartomatoso, con una morfología caracterizada por el crecimiento exagerado y desordenado de células nativas de cada órgano del que provienen; sus características incluyen crecimiento desorganizado de tipo «arboriforme» del músculo liso (muscular de la mucosa) que perforan la lámina propia, y dilatación glandular dentro de la submucosa o muscular propia12; el crecimiento es muy llamativo en los pólipos de ID y menos en los pólipos gástricos y de colon. Los pólipos de ID y colon tienden a peduncularse, mientras que los pólipos de estómago son sésiles12,25,27,28; el epitelio de recubrimiento que se encuentra es el específico del segmento intestinal en el que se encuentra el pólipo.
El aumento de riesgo de presentar cáncer implica el cáncer gastrointestinal (38-66%), el de mama (32-54%), el cáncer de páncreas (11-36%), con menos frecuencia tiroides, pulmón y cáncer en los órganos reproductivos (9-21%)3,27,29,30; se ha descrito que el riesgo absoluto de cáncer de mama llega a ser similar a la magnitud del riesgo observado en las formas hereditarias por mutaciones en la línea germinal de BRCA1 o BRCA229. Los hombres pueden desarrollar tumores únicos testiculares llamados tumores calcificantes de células de Sertoli, que se asemejan a tumores del cordón sexual con túbulos anulares (SCTAT), y también pueden desarrollar ginecomastia y edad ósea avanzada de forma temprana. Los hallazgos ginecológicos asociados con SPJ incluyen metaplasia mucinosa de la trompa de Falopio, adenocarcinoma endocervical y tumores de ovario, como tumores mucinosos quísticos y tumores SCTAT que han sido descritos en niñas de hasta 4 años de edad25,27,31.
El presente estudio trata de realizar una correcta identificación y registro de los pacientes con SPJ, estimar la prevalencia de la enfermedad en la provincia de Valencia, mediante la búsqueda de pacientes con el código CIE-9 ES 759.6 que incluye varias enfermedades no relacionadas entre sí, que son el SPJ, síndrome de Von Hippel Lindau (SVHL), síndrome de Sturger-Weber (SSW) y otras hamartomatosis no clasificadas bajo otros conceptos, por medio de la base de datos del CMBD; de esta forma describiremos algunas de las características de la enfermedad, proporcionaremos una visión de la magnitud de la enfermedad en nuestra área geográfica y además obtendremos los registros adecuados. Hasta la fecha no se ha descrito ninguna herramienta de este tipo ni tampoco se ha publicado ningún trabajo que use esta metodología para estudiar el SPJ en España.
Materiales y métodosDiseño, población e instrumentos del estudioSe realizó un estudio observacional retrospectivo de una serie de casos de la provincia de Valencia (España), que según los datos del Instituto Nacional de Estadística (INE) para el año 2016 contaba con una población de 2,517,951 habitantes32, datos que se utilizaron para estimar la prevalencia. Este trabajo fue aprobado por el Comité de Ética de Investigación Clínica de la Dirección General de Salud Pública de Valencia y de 10 hospitales de la provincia de Valencia; incluimos al Hospital Universitario Doctor Peset Aleixandre, Hospital Universitario y Politécnico La Fe, Hospital General Universitario, Hospital Clínico Universitario, Hospital de Manises, Hospital de Sagunto, Hospital General de Requena, Hospital de la Ribera, Hospital Francesc de Borja de Gandía, Hospital Lluís Alcanyís de Xàtiva e Instituto Valenciano de Oncología.
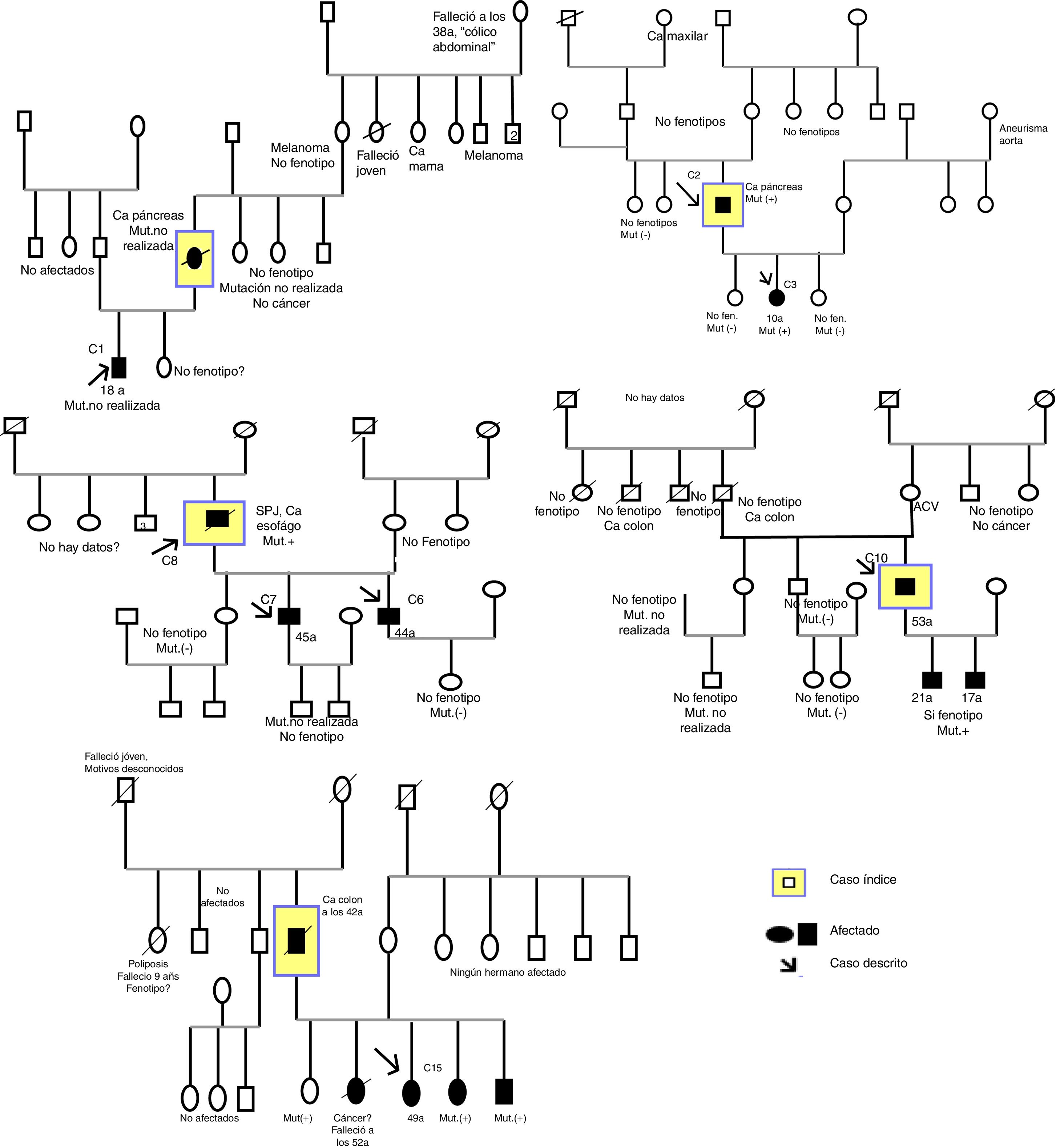
El procedimiento para identificar a los pacientes fue obtener en estos centros hospitalarios un listado de registros informáticos de pacientes incluidos en la base de datos CMBD de ingresos hospitalarios del Ministerio de Sanidad, Política Social e Igualdad de España, desde el 1 de mayo de 1997 hasta el 1 de mayo de 2016, utilizando el sistema de clasificación CIE-9 el código 759.633; de este listado de pacientes se realizó una revisión de cada una de las historias clínicas (HC) con el fin de localizar y clasificar solo a los codificados con el diagnóstico correspondiente a SPJ; adicionalmente se hizo la búsqueda en paralelo de pacientes con el diagnóstico de SPJ que estaban registrados en la base de datos del software informático clínico utilizado en cada hospital, usando las palabras «Peutz-Jeghers», los software informáticos utilizados para esta búsqueda fueron el Orión clinic, Mizar clinic, Iris clinic y Pangea. Posteriormente se solicitó personalmente el consentimiento informado a cada paciente codificado como SPJ. Se procedió a hacer una exhaustiva recogida de datos anonimizados y revisión de las HC, recogiendo los datos basados en un cuestionario realizado para este estudio que incluye diferentes variables clínicas y epidemiológicas de la enfermedad. Se realizaron 10 árboles genealógicos (fig. 1), se señalan los casos incluidos en el estudio, no siempre el caso índice se ha incluido ya que podría no pertenecer a un hospital de la provincia de Valencia o haber fallecido.
: negativo, (+): positivo.")
Además, para asegurar eliminar el sesgo de selección, posterior a la comprobación de los casos con SPJ con los listados iniciales, se confirmó que estuviesen incluidos en la base de datos del Servicio de Promoción de Salud y Prevención en el Entorno Sanitario de la Dirección General de Salud Pública que asume el programa de Cáncer Hereditario en la Comunidad Valenciana.
Es importante recalcar que todos los datos de carácter personal obtenidos en este estudio son confidenciales y se trataron conforme al Reglamento UE 2016/679 del Parlamento Europeo y del Consejo de 27 de abril de 2016 de Protección de Datos.
Criterios de inclusión y exclusiónLos pacientes se incluyeron en el estudio si cumplían uno de los siguientes criterios clínicos: 2 o más pólipos de tipo Peutz Jeghers (PJ) confirmados histológicamente, cualquier número de pólipos de tipo PJ en un paciente con antecedentes familiares de SPJ en un pariente cercano, pigmentación mucocutánea característica en un paciente que tiene historia familiar de SPJ en un pariente cercano, cualquier número de pólipos de tipo PJ en un paciente que tiene pigmentación mucocutánea característica12,34,35.
Se excluyeron los pacientes registrados con el código CIE-9 759.6 que no estaban clasificados como SPJ; dentro de estos encontramos los diagnósticos correspondientes a SSW, SVHL y otras hamartomatosis (pulmonar, mama, cerebral, vesícula biliar, tejidos blandos); además se excluyó a «otros diagnósticos» que erróneamente tenían este código, como son angiomas, adenomas, síndrome de Proteus, síndrome de Cowden, enfermedad de Lhermitte-Duclos y neurofibromatosis.
Descripción de variablesSe recogieron las variables generales como sexo, edad actual, edad de diagnóstico y edad de fallecimiento. Respecto a las variables clínicas se tuvieron en cuenta si tenían o no pigmentación mucocutánea (melanosis) en la boca, la mucosa oral, la nariz, o alrededor, en las manos y los dedos, los pies y los dedos y la región perianal; si presentaron cáncer y su localización, la edad de diagnóstico del cáncer; si era portador o no de pólipos de tipo hamartomatoso y su localización; y si tenían otro tipo de pólipos clasificados como adenomatosos, hiperplásicos o mesenquimales.
En cuanto a las variables relacionadas con los antecedentes familiares se describe la definición de caso familiar cuando 2 o más individuos en una familia estaban afectados; historia familiar positiva (HFP) en un paciente cuando este había referido la presencia de cáncer relacionado con SPJ, algún familiar con SPJ, pigmentación mucocutánea característica y/o poliposis en un familiar de primer grado; y un caso esporádico se define cuando se hizo el diagnóstico clínico o genético en un solo miembro de la familia, siempre y cuando se hubiese descartado en los padres de forma clínica, endoscópica o radiológica la presencia de hamartomas, pigmentación mucocutánea y/o si la mutación es conocida la ausencia de esta, y además que el caso no cumpliera los criterios de tener HFP.
Las variables relacionadas con las complicaciones asociadas son presentación de una primera cirugía gastrointestinal urgente invasiva no endoscópica a lo largo de la evolución de la enfermedad y su edad de presentación, si la había presentado, obstrucción intestinal, invaginación intestinal y su edad de presentación, rectorragia, prolapso de un pólipo, ulceración de un pólipo, anemia, hemorragia digestiva alta (HDA) y perforación intestinal; finalmente, el número de ingresos hospitalarios a lo largo de la evolución de la enfermedad.
Otra de las variables descritas es si el paciente había recibido consejo genético en una Unidad de Consejo Genético en Cáncer Hereditario (UCGCH) mediante una primera visita y/o continua de control y seguimiento en esta unidad en los últimos 10 años.
Finalmente se describió la variable prueba genética, la cual describe si se realizó o no la mutación del gen STK11, su resultado y la descripción de la mutación.
Test genéticoPara obtener los datos de resultados de pruebas de análisis genético se contactó con los diferentes servicios de genética y consejo genético en cáncer hereditario de la provincia de Valencia (Hospital Universitario y Politécnico La Fe, Hospital Universitario Clínico de Valencia e Instituto Valenciano de Oncología), la mayoría de las pruebas se remitieron a la Unidad de Genética Molecular del Hospital General Universitario de Elche, ya que es allí donde se realizan las pruebas genéticas de los pacientes que se incluyen dentro del programa de Cáncer Hereditario de la Comunidad Valenciana36; una prueba se realizó en el Laboratorio de Biología Molecular del Hospital Universitario y Politécnico La Fe, porque en el momento de la realización de la prueba aún no se remitían las muestras al Hospital de Elche.
Análisis estadísticoLos datos se recogieron en una base de datos del software estadístico IBM SPSS Statistics 24.0 y se procesaron con el mismo software estadístico. En el análisis descriptivo se utilizaron las medias y la desviación estándar. Se utilizó la prueba de Levene y prueba de Mann-Whitney para realizar comparación de varianzas y medias de 2 grupos independientes con distribución normal y anormal, respectivamente. Se describieron variables categóricas utilizando frecuencias y porcentajes. Se realizaron los árboles genealógicos con el programa GenoPro 2018.
ResultadosConstrucción y obtención del registro de pacientesSe obtuvo un listado con 405 HC codificadas con CIE-9 759.6, 22 pacientes tenían asignado el diagnóstico de SPJ, pero solo 15 casos cumplían los criterios diagnósticos, de los primeros, uno fue un error de codificación, ya que no tenía ningún síntoma que hiciera sospechar SPJ, uno fue un recién nacido fallecido en el que se describían en la HC antecedentes familiares de SPJ (no fue posible localizar a ninguno de sus familiares), 2 fueron sospechas de SPJ no estudiados y 3 pacientes sospecha de SPJ estudiados y descartados. El C1 pertenece a otra comunidad autónoma y C3 y C11 pertenecen a otra provincia de la Comunidad Valenciana, pero se incluyen en el estudio, ya que han recibido asistencia sanitaria hospitalaria y constan dentro del registro de ingresos hospitalarios de un hospital de la provincia de Valencia (Hospital Universitario y Politécnico La Fe).
Descripción de casos y familias, historia familiar y casos familiares definidoSe describen 11 familias (F) y 15 casos, hay un caso por cada familia en la F5, F6, F7, F8, F9, F10 y en la F2 y F3 hay 2 casos (la F3 se negó a la realización del árbol genealógico). La F11 tiene 5 miembros afectados (padre, 3 hermanas y un hermano), pero solo se incluye un caso (hermana, C15), la F1 tiene 2 casos (madre e hijo), pero solo se incluye un caso (hijo, C1) y la F10 tiene 3 casos afectados (padre y 2 hijos), pero solo se incluye un caso (padre, C10); el motivo por el cual se excluyen algunos casos es porque nunca han acudido a un hospital de la provincia de Valencia y no teníamos acceso a los datos de la HC, ni tampoco están dentro del registro de ingresos hospitalarios de los hospitales de la provincia de Valencia (fig. 1).
Todos los casos son naturales de España, excepto los casos de la F4 que son naturales de Bulgaria.
Para definir un caso como caso familiar definido y un caso con HFP analizamos los árboles genealógicos y los datos clínicos recogidos; los C4 y C5 (F3) se documentan con datos de la HC como casos familiares definidos. Encontramos 10 (67%) casos de 15, que fueron casos familiares definidos y comprendían 6 familias y solo 6 (60%) casos de los 10 tenían HFP (C1, C3, C5, C6, C7, C15); la historia familiar era desconocida en 2 casos y en los otros 2 casos no había HFP. Hay 6/15 casos con HFP y 6/15 (40%) esporádicos, hay 3 casos que no se pueden definir (C4, C8 y C12) porque los antecedentes son desconocidos (tabla 1).
Descripción de los casos
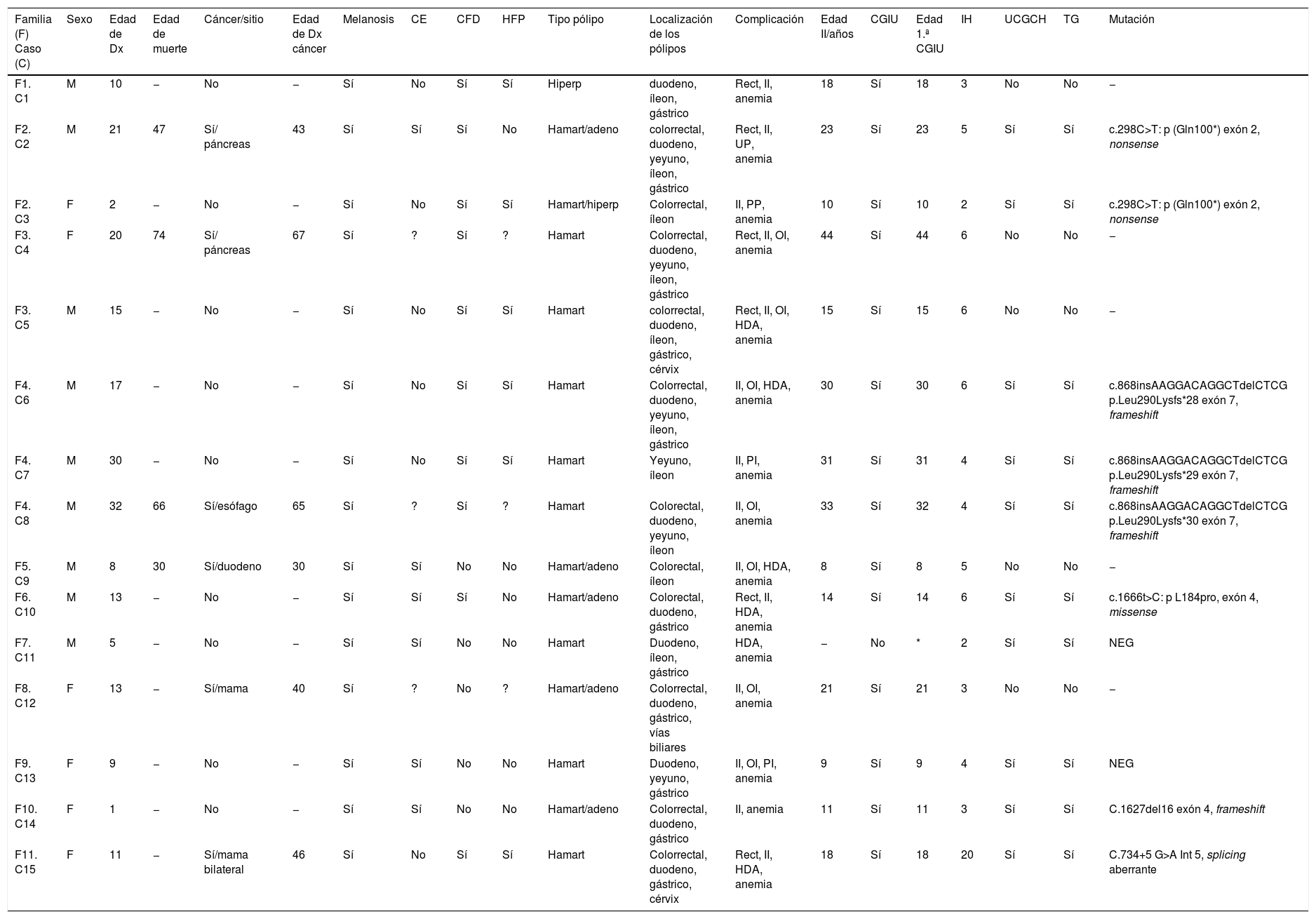
| Familia (F) Caso (C) | Sexo | Edad de Dx | Edad de muerte | Cáncer/sitio | Edad de Dx cáncer | Melanosis | CE | CFD | HFP | Tipo pólipo | Localización de los pólipos | Complicación | Edad II/años | CGIU | Edad 1.ª CGIU | IH | UCGCH | TG | Mutación |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F1. C1 | M | 10 | − | No | − | Sí | No | Sí | Sí | Hiperp | duodeno, íleon, gástrico | Rect, II, anemia | 18 | Sí | 18 | 3 | No | No | − |
| F2. C2 | M | 21 | 47 | Sí/ páncreas | 43 | Sí | Sí | Sí | No | Hamart/adeno | colorrectal, duodeno, yeyuno, íleon, gástrico | Rect, II, UP, anemia | 23 | Sí | 23 | 5 | Sí | Sí | c.298C>T: p (Gln100*) exón 2, nonsense |
| F2. C3 | F | 2 | − | No | − | Sí | No | Sí | Sí | Hamart/hiperp | Colorrectal, íleon | II, PP, anemia | 10 | Sí | 10 | 2 | Sí | Sí | c.298C>T: p (Gln100*) exón 2, nonsense |
| F3. C4 | F | 20 | 74 | Sí/ páncreas | 67 | Sí | ? | Sí | ? | Hamart | Colorrectal, duodeno, yeyuno, íleon, gástrico | Rect, II, OI, anemia | 44 | Sí | 44 | 6 | No | No | − |
| F3. C5 | M | 15 | − | No | − | Sí | No | Sí | Sí | Hamart | colorrectal, duodeno, íleon, gástrico, cérvix | Rect, II, OI, HDA, anemia | 15 | Sí | 15 | 6 | No | No | − |
| F4. C6 | M | 17 | − | No | − | Sí | No | Sí | Sí | Hamart | Colorrectal, duodeno, yeyuno, íleon, gástrico | II, OI, HDA, anemia | 30 | Sí | 30 | 6 | Sí | Sí | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*28 exón 7, frameshift |
| F4. C7 | M | 30 | − | No | − | Sí | No | Sí | Sí | Hamart | Yeyuno, íleon | II, PI, anemia | 31 | Sí | 31 | 4 | Sí | Sí | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*29 exón 7, frameshift |
| F4. C8 | M | 32 | 66 | Sí/esófago | 65 | Sí | ? | Sí | ? | Hamart | Colorectal, duodeno, yeyuno, íleon | II, OI, anemia | 33 | Sí | 32 | 4 | Sí | Sí | c.868insAAGGACAGGCTdelCTCG p.Leu290Lysfs*30 exón 7, frameshift |
| F5. C9 | M | 8 | 30 | Sí/duodeno | 30 | Sí | Sí | No | No | Hamart/adeno | Colorectal, íleon | II, OI, HDA, anemia | 8 | Sí | 8 | 5 | No | No | − |
| F6. C10 | M | 13 | − | No | − | Sí | Sí | Sí | No | Hamart/adeno | Colorectal, duodeno, gástrico | Rect, II, HDA, anemia | 14 | Sí | 14 | 6 | Sí | Sí | c.1666t>C: p L184pro, exón 4, missense |
| F7. C11 | M | 5 | − | No | − | Sí | Sí | No | No | Hamart | Duodeno, íleon, gástrico | HDA, anemia | − | No | * | 2 | Sí | Sí | NEG |
| F8. C12 | F | 13 | − | Sí/mama | 40 | Sí | ? | No | ? | Hamart/adeno | Colorrectal, duodeno, gástrico, vías biliares | II, OI, anemia | 21 | Sí | 21 | 3 | No | No | − |
| F9. C13 | F | 9 | − | No | − | Sí | Sí | No | No | Hamart | Duodeno, yeyuno, gástrico | II, OI, PI, anemia | 9 | Sí | 9 | 4 | Sí | Sí | NEG |
| F10. C14 | F | 1 | − | No | − | Sí | Sí | No | No | Hamart/adeno | Colorrectal, duodeno, gástrico | II, anemia | 11 | Sí | 11 | 3 | Sí | Sí | C.1627del16 exón 4, frameshift |
| F11. C15 | F | 11 | − | Sí/mama bilateral | 46 | Sí | No | Sí | Sí | Hamart | Colorrectal, duodeno, gástrico, cérvix | Rect, II, HDA, anemia | 18 | Sí | 18 | 20 | Sí | Sí | C.734+5 G>A Int 5, splicing aberrante |
ADC: adenocarcinoma; Adeno: adenomatoso; CE: caso esporádico; CFD: caso familiar definido; CGIU: cirugía gastrointestinal urgente; Dx: diagnóstico; F: femenino; GI: gastrointestinal; Hamart: hamartomatoso; HDA: hemorragía digestiva alta; HFP: historia familiar positiva; Hiperp: hiperplásico; IH: ingresos hospitalarios; II: invaginación intestinal; M: masculino; OI: obstrucción intestinal; PI: perforación intestinal; PP: prolapso pólipo; Rect: rectorragia; TG: test genético; UCGCH: unidad de consejo genético en cáncer hereditario; UP: ulceración pólipo; ?: desconocido.
Se realizó el test genético a 7 de las 11 familias y a 10 de los 15 casos, identificando la mutación en 5 familias (71%) y en 8 casos (80%), el C10 fue inicialmente una variante con efecto clínico o significado desconocido, pero que en la actualidad según la revisión de las bases de datos podría ser considerada como una variante probablemente patogénica; solo el C11 y C13 (20%) fueron negativos.
De los 6 casos con HFP se les hizo la prueba genética a 4 de ellos, identificando en todos una mutación patogénica, no se solicitó test genético al C1 y el C5 no quiso realizárselo.
Las mutaciones encontradas se observan en la tabla 1. Hay 5 mutaciones, una (20%) de tipo nonsense (F2), una (20%) missense (F6), 2 (40%) frameshift (F4 y F10) y una (20%) de tipo splicing aberrante (F11).
Características epidemiológicasDe los 15 casos 9 son hombres (60%) y 6 mujeres (40%), con una relación hombre:mujer 1.5:1. La edad rango de participantes fue entre 10 y 60 años. La prevalencia puntual estimada para el año 2016 fue de 0.4/100,000 habitantes.
El rango de edad de diagnóstico fue entre uno y 32 años, con una edad media de diagnóstico de 13.8±9.08 años; en hombres de 17.3±9.40 años, y en mujeres de 8.5±5.78 años (p=0.062). La edad media de fallecimiento fue de 54.2±19.7 años.
Presentación clínica y complicacionesTodos los pacientes tenían el fenotipo de hiperpigmentación mucocutánea y eran portadores de pólipos, pero en uno de ellos (C1) no eran hamartomatosos; este caso era portador de pólipos hiperplásicos, aunque cumplía criterios para SPJ, además, 5 casos presentaron pólipos adenomatosos, diagnosticados entre los 13 y 43 años (media de edad 31±15.5 años) después del diagnóstico de SPJ. Cabe destacar que el C2 y C9 con pólipos adenomatosos presentaron cáncer de páncreas y de ID (duodenal) respectivamente. Solo el C3 presentó un pólipo hiperplásico a los 7 años después del diagnóstico y no tuvo ningún tipo de cáncer.
En la localización de los pólipos todos los casos tenían pólipos en el ID (80% duodeno, 67% íleon y 40% en yeyuno), 11 casos (73.3%) tenían pólipos colorrectales y 11 (73.3%) gástricos; destacamos que todos los casos eran portadores de pólipos en más de una localización. Se reportaron 3 (20%) casos portadores de pólipos extraintestinales (C12 en la vesícula biliar, C5 y C15 en el cérvix uterino).
Todos los casos han tenido una complicación gastrointestinal, de los cuales 14 (93.3%) han tenido una invaginación intestinal (edad media de 19±10.3 años), 9 (60%) han tenido obstrucción intestinal (edad media de 25±11.5 años), 6 (40%) rectorragia (edad media de 26±12.3 años), 6 (40%) HDA (edad media de 23±9.7 años), uno (6.7%) ha tenido prolapso de un pólipo (a los 2 años), otro (6.7%) ulceración de un pólipo (a los 30 años) y todos los casos han tenido anemia; 14 casos (93.3%) han requerido una primera CGUI (edad media de 19±10.2 años) y una media de ingresos hospitalarios de 5±4.3 a lo largo de la evolución de la enfermedad.
Presentación de cáncerDe todos los pacientes 6 casos (40%) presentaron cáncer, con una edad media de diagnóstico de cáncer de 48.5±14.60 años (rango de 30 a 67 años), 4 (67%) son hombres y 2 (33%) son mujeres. Según la edad de diagnóstico de cáncer el C9 fue el que presento cáncer de forma más temprana. La causa de los 4 fallecimientos fue por cáncer. El 13% de los casos presentó cáncer de páncreas con una edad media de 55 años. De los 8 casos con la mutación 3 desarrollaron cáncer (37.5%). Los tumores más frecuentes fueron mama y páncreas. Se describen las características, tipo histológico del cáncer y edad de diagnóstico en la tabla 1.
Consejo genético en cáncer hereditarioHan acudido a una UCGCH 10 casos (67%), y los 5 (33%) que no acudieron fueron los C4 y C9 que fallecieron hace más de 10 años, el C1 porque no ha sido remitido, el C5 por motivos desconocidos y el C12 porque no deseaba acudir.
DiscusiónEn algunos países los datos de los registros de pacientes con SPJ están disponibles gracias a los registros nacionales de poliposis37. Uno de los registros más antiguos del mundo de poliposis es el Registro del Hospital de St. Mark en Londres16. Cabe destacar a Tan et al. (2010) en su estudio realizado en Singapur26, identificando de 610 casos del registro de poliposis a solo 7 pacientes con SPJ. En España no hay reportado ningún registro de poliposis, aunque sí existe el específico para la poliposis adenomatosa familiar38.
Como en este trabajo, Fostira et al. (2018), en su estudio de 8 pacientes griegos, y Tchekmedyian et al. (2013), en su estudio con 25 casos uruguayos, hablan y describen la creación de un registro de pacientes con SPJ30,39. Existen descritos artículos académicos de estudios observacionales retrospectivos realizados a nivel mundial que utilizan diferentes metodologías para la identificación de pacientes, algunos utilizan bases de datos de laboratorios clínicos y el test genético para STK11, otros emplean bases de datos de los laboratorios de histopatología relacionados con poliposis hamartomatosa o cáncer, pero en concreto no existe descrita una metodología precisa para obtener un registro adecuado de pacientes14,25,38.
Esta enfermedad necesita de forma precisa y consensuada una nomenclatura y una clasificación; se han creado diferentes clasificaciones, por ejemplo, la red y base de datos de ER y medicamentos huérfanos Orphanet40,41, clasifica al SPJ con el número 2,869, pero dentro de varias subcategorías y grupos; en esta base de datos se incluyen otras codificaciones como la descrita por Online Mendelian Inheritance in Man® que asignan al SPJ el #175,200, la cual se centra en la relación entre el fenotipo y el genotipo; además, incluyen los códigos MeSH D010580 y MedDRA 10034764, terminología médica normalizada para intercambiar información. Por otro lado, los anatomopatólogos crean a nivel internacional un sistema de nomenclatura llamada SNOMED-CT, de uso restringido; este sistema fue utilizado con el código S54320 por el grupo de Jelsig et al. (2016) para la identificación de los casos en el estudio realizado en 46 pacientes daneses20.
Desde hace más de 20 años en España se utilizaba el sistema de codificación CIE-933, que recoge aproximadamente 100 códigos para ER; la Unión Europea recomendó el desarrollo del inventario de ER dentro una nueva implantación del sistema de codificación CIE-1042; esta versión de clasificación CIE-10 incluye unos 300 códigos, correspondientes a ER. Esta última versión (CIE-10) ofrecía más posibilidades de conseguir catalogar y clasificar una ER, y así facilitaría el estudio grupal de ER relacionadas entre sí1, pero en el caso del SPJ CIE-10 ES utiliza el código Q85.8 denominado «otras facomatosis no clasificadas bajo otro concepto» y agrupa las mismas 3 enfermedades descritas en el anterior código CIE-9 (SPJ, SSW y SVHL), las cuales no están relacionadas entre sí; al menos, sí que se consigue excluir el diagnóstico de «otras hamartomatosis no clasificadas bajo otros conceptos», afirmando de esta forma que en la búsqueda no habrá listados con gran número de pacientes codificados erróneamente e incluidos dentro de una clasificación general como lo es «otras hamartomatosis», y se deja de englobar un sin fin de diagnósticos de hamartomas en diferentes sitios anatómicos, sino que además descartaremos otras ER que tienen un código específico diferente. Actualmente en España se está realizando una conversión de todos los códigos CIE-9 a códigos CIE-10, y en el caso de las ER también se construyen bases de datos y registros, intentado consensuar el uso común de códigos Online Mendelian Inheritance in Man®, Orpha y CIE-10.
Nosotros utilizamos en este estudio la codificación CIE-9, ya que en el periodo estudiado aún se codificaba con CIE-9 y en el momento de la recogida de datos no estaban totalmente reclasificados y convertidos a CIE-10. A pesar de que la clasificación CIE-9 es poco específica, se encontró, validó y realizó una descripción clínica de los casos de la provincia de Valencia.
Nuestra metodología fue utilizar la base de datos del CMBD, ya que muchos de nuestros pacientes acuden a hospitales de alto nivel, necesitando atención en el servicio de urgencias y en algunas ocasiones requieren ingresos intrahospitalarios, sea por complicaciones graves sea porque necesitan de pruebas complementarias o tratamientos intrahospitalarios especiales1. Sin embargo, la experiencia de este estudio dice que para poder realizar la validación y seguimiento de pacientes con SPJ se revisaron un gran número de HC para solo obtener una n de 15 pacientes. Podemos defender la idea de que la poca prevalencia de esta ER justifica que la n del estudio sea baja, pero no podemos dejar pasar por alto que al no contar con otro sistema de codificación y clasificación más específico en este momento para el registro de esta ER, no hay otra forma precisa de cribar y registrar a estos pacientes, y que el uso de este sistema de clasificación hace que las búsquedas y validaciones epidemiológicas de las ER requieran de un largo y quizá poco exitoso trabajo. No obstante, al parecer, en otros lugares del mundo también utilizan la clasificación CIE-9 para la búsqueda de pacientes, por ejemplo en un estudio americano realizado en niños usan como herramienta de búsqueda la clasificación CIE-943. Es interesante nombrar a Jelsig et al., que de una base de registro nacional de pacientes daneses utilizó el código CIE-10 DQ858B, pero, en este registro, utilizan extensiones y añaden A, B, C para códigos de enfermedades específicas, como es el caso del SPJ, que a su código CIE-10 internacional se le añade una B, de esta forma el registro de una ER, como es el SPJ, es más específico20,44.
Cabe destacar un posible sesgo de selección como una limitación significativa del presente estudio, debido a que es una revisión retrospectiva que incluye a los pacientes que han acudido a un hospital, por lo tanto, es posible que los niños asintomáticos no estén en este registro y se hayan quedado excluidos del estudio.
Las cifras descritas para la prevalencia de la enfermedad son variables; en las múltiples revisiones bibliográficas utilizan diferentes metodologías para su cálculo. En los numerosos artículos de revisión hablan de prevalencias desde 1:50,000 a 1:280,00020,30,45–47. Los datos proporcionados por Orphanet describen en su informe N.2 del año 2017 una prevalencia estimada en Europa de 0.4 en 100,000 nacimientos48. Se habla de incidencias desde 1:8,300 a 1:200,000 nacimientos3,12,13,18,35,39,49. La prevalencia estimada en nuestra serie fue de 0.4/100,000 habitantes para el año 2016, la cual se encuentra dentro de los rangos descritos dentro de otras publicaciones.
Se han reportado edades medias de diagnóstico de 17 a 26 años17,26,39,50–52. La edad media de diagnóstico de esta serie comprende una edad pediátrica; en los hombres es casi el doble que en las mujeres, sin embargo, no se encontraron diferencias estadísticamente significativas. No se ha descrito predominio de sexo5,46,47, ni por raza17; se habla de una ratio hombre:mujer en el estudio uruguayo de 1:0.6739, concordante con nuestros resultados, contrariamente Tan et al. describen un 1:2.526.
La edad media de fallecimiento de esta serie es 5 a 10 años mayor a lo reportado en el estudio danés, que fue de 49.5 años, y a la del estudio uruguayo con 44 años20,39. Este último estudio coincide con los datos observados en nuestra población, en los que la causa más frecuente de muerte fue el cáncer, con una edad media de fallecimiento similar39. Todo lo anterior nos hace pensar que los pacientes con SPJ fallecen en un rango de edad joven.
Inicialmente, en el año 1996 el grupo de Tomlinson descartó probables locus en el cromosoma 1 y 64; posteriormente Hemminki et al. (1998)11 describieron la mutación causante en línea germinal del gen STK11, el cual tiene una longitud de 23Kb, contiene 9 exones codificantes para una proteína serina treonina quinasa de 433 aminoácidos (Lkb1) y un exón final no codificante. La región codificante o catalítica (dominio proteína quinasa) se encuentra entre los aminoácidos 44 y 309; las regiones C-terminales y N- terminales no tienen analogía con otras quinasas de la misma familia. La proteína Lkb1 se expresa en todos los tejidos humanos, aunque con mayor intensidad en testículos e hígado fetal, y se encuentra en el núcleo y citoplasma de la célula. Se ha sugerido la probable existencia de otro locus en 19q1313,53,54.
Existen más de 300 mutaciones humanas en STK11 en la base de datos NCBI (https://www.ncbi.nlm.nih.gov/clinvar)55. Se ha descrito que aproximadamente el 85% son pequeñas mutaciones, y de estas el 35% son mutaciones puntuales que incluyen las de tipo missense y nonsense, 50% son inserciones pequeñas y deleciones y el 15% que afectan los sitios de splicing56,57, siendo esto concordante con nuestros resultados, siendo el 40%, 40% y 20% respectivamente. Actualmente los 5 tipos de mutaciones halladas en nuestro estudio se han descrito en las bases de datos.
Se ha descrito que una mutación puede estar en hasta 50-94% de los casos afectados8,12,25,30,50,58. Se encontró en nuestra serie un porcentaje alto de casos (80%) y familias (71%) con el test genético realizado que tenían una mutación, coincidiendo con nosotros el estudio danés y el realizado por el grupo de Fostira et al., reportando una tasa y prevalencia de mutación del 86% y 87.5% respectivamente20,30; en cambio, el estudio realizado por Scott et al.(2002) en 14 pacientes australianos describió la mutación en menos del 50% de todos los pacientes, y el estudio realizado en Estados Unidos por Amos et al. (2004), con una serie de 51 pacientes, describió la mutación en el 69% de los probandos51,59. Anteriormente se pensaba que la mutación se encontraba en la mayoría de pacientes, pero McGarrity et al. (2000) propusieron que la mutación no era responsable en todos los SPJ, ya que ellos la identificaron en el 60% de los casos familiares y en el 50% de los casos esporádicos. Esto puede ser explicado por el uso de diferentes métodos de análisis mutacional, por la posibilidad de heterogeneidad del locus y de la enfermedad o, porque en realidad, sí que existe un segundo gen responsable del SPJ.
Se reporta que hasta un 25-30% de los casos de SPJ son esporádicos, posiblemente por mutaciones de novo o por variantes de baja penetrancia6,24. Se han identificado mutaciones en el 50-90% de los casos esporádicos13,28,51. En nuestra serie el porcentaje de casos esporádicos es un 10-15% más alto que lo descrito (tabla 1); esto podría ser explicado porque en la mayoría de estudios dentro de la definición de caso esporádico incluyen la realización de un test genético al caso y que el de sus padres sea negativo13,50, pero nosotros no incluimos esta condición para definir un caso esporádico, ya que años atrás en nuestra población no contábamos con la disponibilidad actual de realizar un test genético, y antes del año 2008 no existía un protocolo para el SPJ incluido en los programas de cáncer hereditario dentro del sistema sanitario valenciano.
En múltiples estudios se reporta que del 36-50% de los casos tienen HFP, coincidiendo con nuestra serie20,22,26,30,39,51 (tabla 1).
Se ha demostrado en varios estudios que de las características clínicas más frecuentes y típicas son la presentación de máculas hiperpigmentadas, así mismo se demuestra en nuestro estudio13,26,39,50.
Se describe que del 80-100% de los pacientes tienen poliposis en el tracto gastrointestinal; en nuestra serie todos los pacientes tenían pólipos, se encontraron además pólipos de tipo hiperplásico y adenomatoso, y así mismo se documentó en el estudio danés20. De los casos portadores de un segundo pólipo adenomatoso uno presentó cáncer de ID; esto puede asociarse a lo descrito en la literatura, que habla de que puede haber transformación de una porción de pólipos hamartomatosos en tumores adenomatosos y posterior carcinoma22,60,61. Bardeesy et al. (2002) sugirieron que la pérdida de expresión del gen STK11 en el epitelio da como resultado pólipos benignos, y que la pérdida de expresión en una lesión en estadio tardío facilita el desarrollo de lesiones con un potencial maligno62.
Los pólipos pueden aparecer en otros sitios como las fosas nasales, los bronquios, el tracto biliar, el uréter y la vejiga, el cérvix uterino y la vagina12,20,25,27,39,63, así como en nuestra serie que se encontraron 3 pacientes con pólipos extraintestinales.
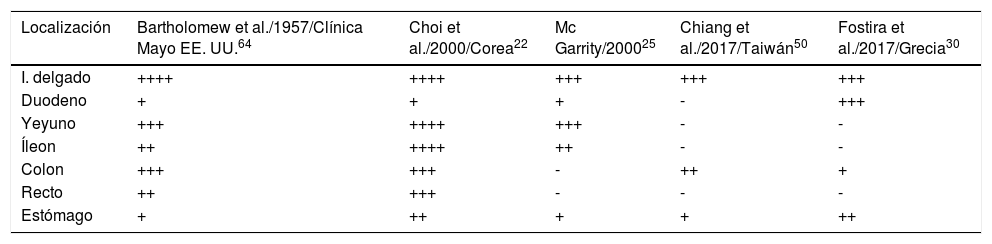
Los pólipos ocurren más frecuentemente en el ID (64-96%)26,39, así mismo ocurre en nuestra serie donde todos los casos tienen pólipos en el ID, seguido del estómago y el colon, en concordancia con el estudio taiwanés50. En la tabla 2 se resumen las localizaciones descritas como más frecuentes de los pólipos. De los localizados en el ID informan que los de mayor frecuencia son los del yeyuno, en contradicción con nuestros resultados que indican que son más frecuentes en el duodeno22,25,26,64.
Localizaciones más frecuentes de los pólipos en pacientes con SPJ
| Localización | Bartholomew et al./1957/Clínica Mayo EE. UU.64 | Choi et al./2000/Corea22 | Mc Garrity/200025 | Chiang et al./2017/Taiwán50 | Fostira et al./2017/Grecia30 |
|---|---|---|---|---|---|
| I. delgado | ++++ | ++++ | +++ | +++ | +++ |
| Duodeno | + | + | + | - | +++ |
| Yeyuno | +++ | ++++ | +++ | - | - |
| Íleon | ++ | ++++ | ++ | - | - |
| Colon | +++ | +++ | - | ++ | + |
| Recto | ++ | +++ | - | - | - |
| Estómago | + | ++ | + | + | ++ |
++++: muy frecuente, -: no reportado.
Las complicaciones más frecuentes que se asocian a los pólipos generalmente son quirúrgicas y urgentes; aproximadamente el 50% de los pacientes presenta síntomas abdominales antes de los 20 años, particularmente secundarios a obstrucción intestinal e invaginaciones17; las invaginaciones generalmente son causadas por pólipos>15mm y el tratamiento principalmente es quirúrgico65. Los pacientes con SPJ tienen un riesgo acumulado alto de presentar invaginaciones intestinales a edades tempranas. Van Lier et al. (2011), en su serie de 110 casos holandeses, reportan que el riesgo de invaginación intestinal fue del 15% (IC del 95% ±8-22%) a los 10 años, 50% (±40-60%) a los 20 años, 65% (±55-75%) a la edad de 30 años, 77% (±68-86%) a la edad de 40 años y 84% (±75-93%) a la edad de 50 años65. Nuestra serie concluye que la anemia y la invaginación intestinal son las complicaciones más frecuentes, seguidas de obstrucción intestinal, rectorragia, HDA, prolapso de un pólipo y ulceración de un pólipo. Coinciden con nuestros resultados el estudio en 30 pacientes coreanos descrito por el grupo de Choi et al. (2000), el taiwanés, el uruguayo y el holandés, los cuales reportan que la invaginación es una de las complicaciones más frecuentes y que es la mayor causa de cirugía abdominal urgente. Las edades medias de invaginaciones descritas son de 16 a 20 años, en relación con la nuestra que fue de 19 años22,39,50,65, aunque existen descritas otras series de niños que describen una edad mediana de presentación de invaginación a los 10 años. En esta misma serie reportan que el 40% de los 14 pacientes pediátricos con SPJ ya habían desarrollado complicaciones a la edad de 8 años, incluyendo invaginación intestinal y rectorragia43. Destacamos en nuestra serie que la mayoría de los casos ha requerido una primera cirugía gastrointestinal urgente invasiva con una edad media de 19 años y la temprana edad de presentación de las complicaciones e intervenciones quirúrgicas. Esto podría ser debido a un déficit intrínseco o extrínseco en el control y/o seguimiento de los pólipos, ya que los pólipos son la principal causa de invaginaciones.
En el caso de la anemia esta es una causa de morbilidad significativa, ya que todos nuestros pacientes la padecieron, pero no se puede relacionar con una etiología concreta, a pesar de que el 40% de casos presentaron rectorragias y el 40% hemorragia digestiva alta. En los otros pacientes no se puede descartar sangrado intestinal microscópico, ya que en las HC no constaba de forma fiable la realización seriada de test de sangre oculta en heces. Tan et al.26, hacen una relación de hemorragia gastrointestinal con la anemia y hablan de que esta situación confería morbilidad en el 42% (n=3) de los casos. Inferior a nuestros resultados, el estudio uruguayo describe que solo la mitad de los pacientes presentó anemia39.
No se encontró bibliografía que destaque el número de ingresos hospitalarios de los pacientes con SPJ para relacionarlo con nuestro trabajo.
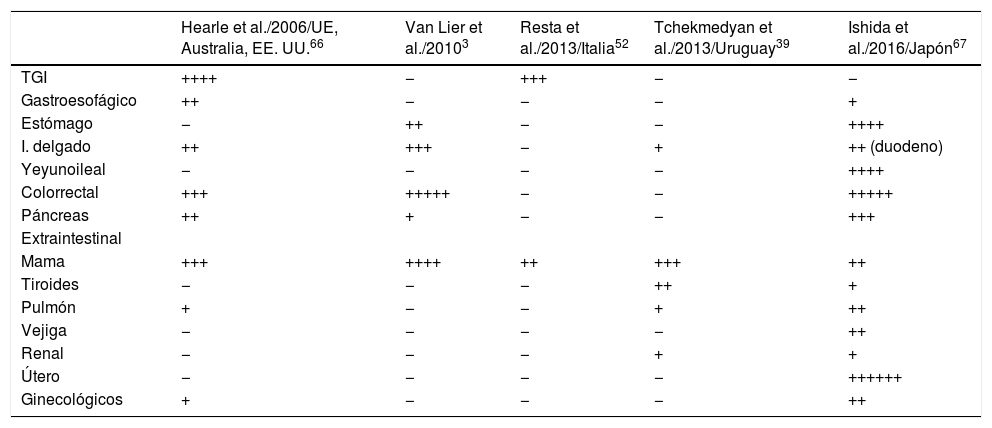
En la tabla 3 se describen los sitios más frecuentes de cáncer en pacientes con SPJ3,5,39,52,66,67. En esta serie los tumores más frecuentes fueron mama y páncreas. Entre los cánceres no gastrointestinales se han descrito tumores de mama, endometrio, cérvix, ovario, pulmón, vesícula biliar13, tiroides20,39, nasosinusales, de pene, renales y de próstata50,68,69. Además se ha informado de la aparición de otros tipos de tumores, como los SCTAT en el ovario, y tumores testiculares de tipo sexual y de células calcificantes de Sertoli, con una edad media de presentación de cáncer de testículo de 9 años (IC 95%: 4-13) y un rango de 3 a 20 años29. En nuestra serie no fueron diagnosticados estos últimos tipos de tumores.
Frecuencia de aparición de tumores en pacientes con SPJ
| Hearle et al./2006/UE, Australia, EE. UU.66 | Van Lier et al./20103 | Resta et al./2013/Italia52 | Tchekmedyan et al./2013/Uruguay39 | Ishida et al./2016/Japón67 | |
|---|---|---|---|---|---|
| TGI | ++++ | − | +++ | − | − |
| Gastroesofágico | ++ | − | − | − | + |
| Estómago | − | ++ | − | − | ++++ |
| I. delgado | ++ | +++ | − | + | ++ (duodeno) |
| Yeyunoileal | − | − | − | − | ++++ |
| Colorrectal | +++ | +++++ | − | − | +++++ |
| Páncreas | ++ | + | − | − | +++ |
| Extraintestinal | |||||
| Mama | +++ | ++++ | ++ | +++ | ++ |
| Tiroides | − | − | − | ++ | + |
| Pulmón | + | − | − | + | ++ |
| Vejiga | − | − | − | − | ++ |
| Renal | − | − | − | + | + |
| Útero | − | − | − | − | ++++++ |
| Ginecológicos | + | − | − | − | ++ |
+++++: muy frecuente; −: no reportado; EE. UU.: Estados Unidos de América; ginecológicos: útero, ovario, cérvix; TGI: tracto gastrointestinal (colon, intestino delgado, esófago, estómago y páncreas); UE: Unión Europea.
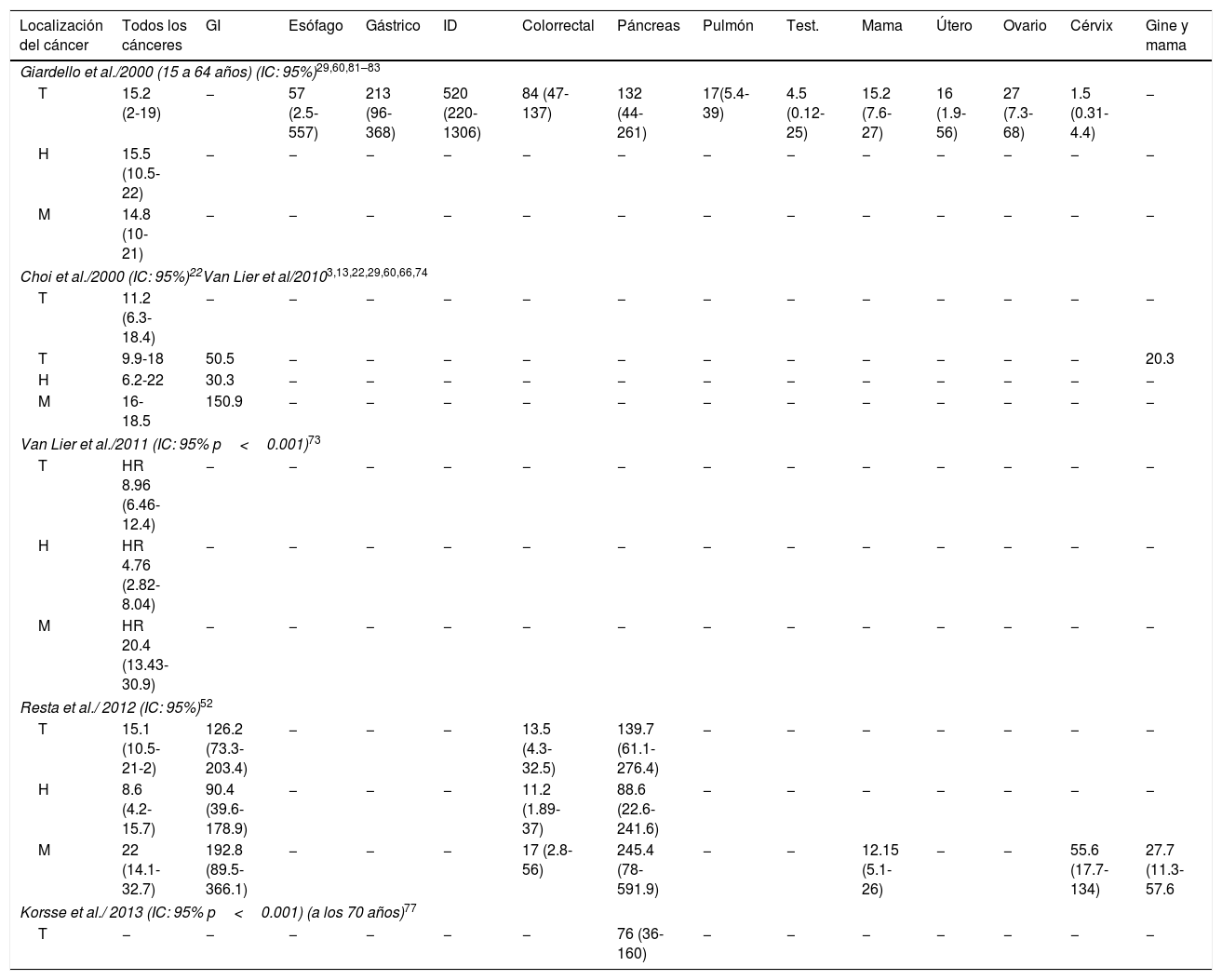
Además del control y vigilancia de los pólipos, otro de los objetivos de los protocolos de seguimiento del SPJ es la realización de un screening y detección precoz de cáncer, ya que desde los años 60 se han descrito casos de cáncer en pacientes con SPJ61,70–72. Se han publicado múltiples estudios que utilizan diferentes metodologías, cuyo objetivo ha sido demostrar que los pacientes con SPJ tienen un elevado riesgo de padecer cáncer de tipo gastrointestinal y extraintestinal, y en estos se reportan los riesgos acumulados (RA) y los riesgos relativos (RR) de padecer cáncer60. En la tabla 4 se realiza una comparación de los diferentes RR descritos. Todos los autores coinciden en describir un RR para desarrollar todo tipo de cáncer dentro de un rango de 9.9 a 18. Giardello et al. (2000), en un metaanálisis, también observan un aumento estadísticamente significativo de RR para cáncer de esófago, estómago, ID, colon, páncreas, pulmón, mama, útero y ovario, y Resta et al. hablan de riesgos relativamente elevados para los cánceres gastrointestinales y ginecológicos en mujeres, y en particular para el cáncer de páncreas y el de cérvix uterino. Todos los autores coinciden en reportar RR mayores en las mujeres que en los hombres, aunque en nuestro estudio se encontraron más diagnósticos de cáncer en los hombres que en las mujeres3,22,29,52,73.
Riesgos relativos de cáncer reportados en pacientes con SPJ
| Localización del cáncer | Todos los cánceres | GI | Esófago | Gástrico | ID | Colorrectal | Páncreas | Pulmón | Test. | Mama | Útero | Ovario | Cérvix | Gine y mama |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Giardello et al./2000 (15 a 64 años) (IC: 95%)29,60,81–83 | ||||||||||||||
| T | 15.2 (2-19) | − | 57 (2.5-557) | 213 (96-368) | 520 (220-1306) | 84 (47-137) | 132 (44-261) | 17(5.4-39) | 4.5 (0.12-25) | 15.2 (7.6-27) | 16 (1.9-56) | 27 (7.3-68) | 1.5 (0.31-4.4) | − |
| H | 15.5 (10.5-22) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| M | 14.8 (10-21) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Choi et al./2000 (IC: 95%)22Van Lier et al/20103,13,22,29,60,66,74 | ||||||||||||||
| T | 11.2 (6.3-18.4) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| T | 9.9-18 | 50.5 | − | − | − | − | − | − | − | − | − | − | − | 20.3 |
| H | 6.2-22 | 30.3 | − | − | − | − | − | − | − | − | − | − | − | − |
| M | 16-18.5 | 150.9 | − | − | − | − | − | − | − | − | − | − | − | − |
| Van Lier et al./2011 (IC: 95% p<0.001)73 | ||||||||||||||
| T | HR 8.96 (6.46-12.4) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| H | HR 4.76 (2.82-8.04) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| M | HR 20.4 (13.43-30.9) | − | − | − | − | − | − | − | − | − | − | − | − | − |
| Resta et al./ 2012 (IC: 95%)52 | ||||||||||||||
| T | 15.1 (10.5-21-2) | 126.2 (73.3-203.4) | − | − | − | 13.5 (4.3-32.5) | 139.7 (61.1-276.4) | − | − | − | − | − | − | − |
| H | 8.6 (4.2-15.7) | 90.4 (39.6-178.9) | − | − | − | 11.2 (1.89-37) | 88.6 (22.6-241.6) | − | − | − | − | − | − | − |
| M | 22 (14.1-32.7) | 192.8 (89.5-366.1) | − | − | − | 17 (2.8-56) | 245.4 (78-591.9) | − | − | 12.15 (5.1-26) | − | − | 55.6 (17.7-134) | 27.7 (11.3-57.6 |
| Korsse et al./ 2013 (IC: 95% p<0.001) (a los 70 años)77 | ||||||||||||||
| T | − | − | − | − | − | − | 76 (36-160) | − | − | − | − | − | − | − |
GI: gastrointestinal (colorrectal, intestino delgado, gástrico, esófago, páncreas); Gine: ginecológico; H: hombre; IC: intervalo de confianza; ID: intestino delgado; M: mujer; T: total; Test.: testículo.
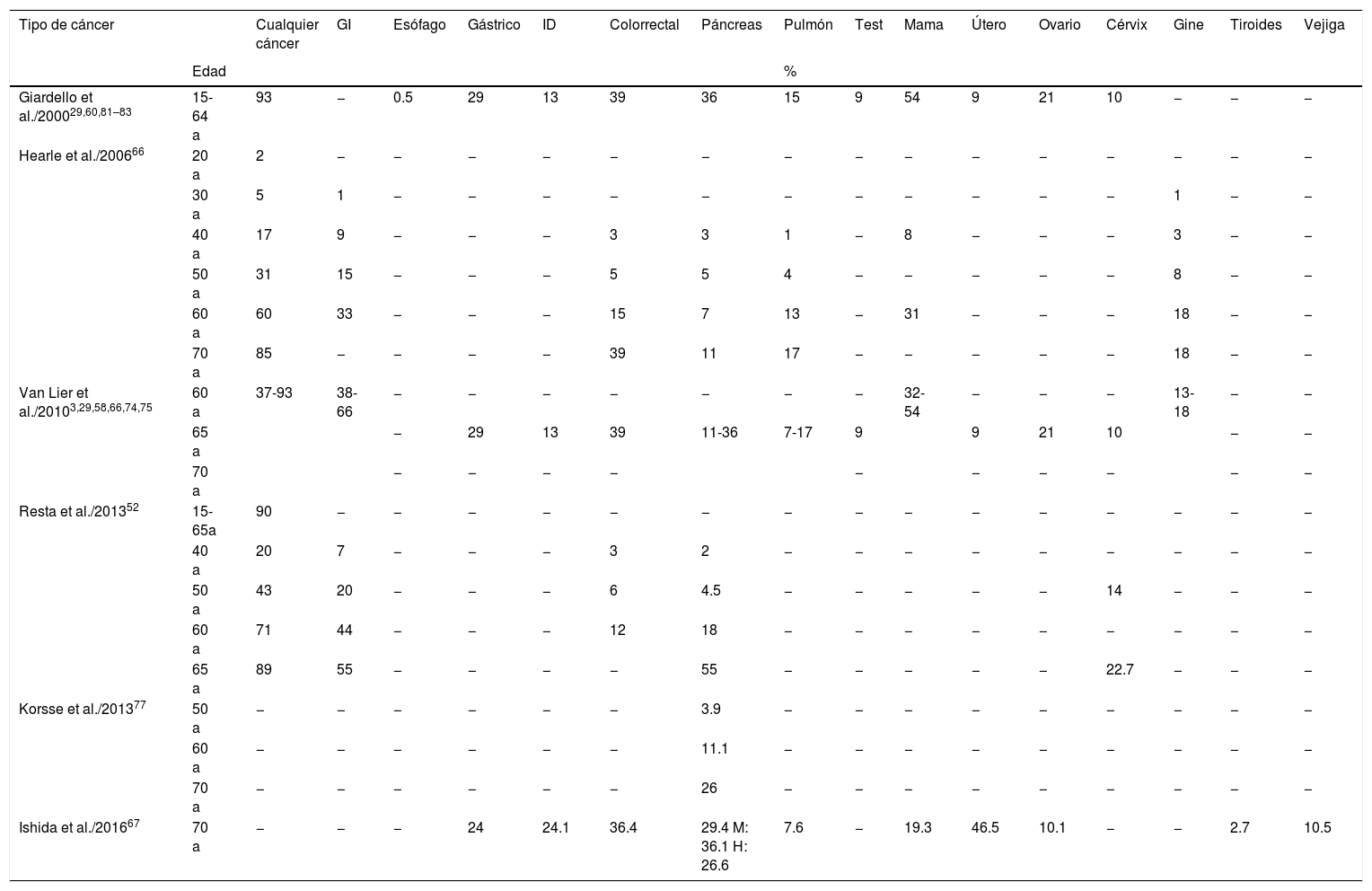
En la tabla 5 describimos los RA por edades; vale la pena resaltar que se reportan RA para todos los tipos de cáncer con una edad de 60 a 70 años del 60% al 93%, para cáncer colorrectal del 15% al 39%, cáncer gastrointestinal del 33% al 66% y de mama del 19% al 54%. Hearle et al. (2006), en su serie de 419 casos, describen que en mujeres el riesgo de cáncer de mama aumenta de forma importante, siendo del 8% y el 31% a las edades de 40 y 60 años respectivamente3,29,52,58,66,67,73–75. Podemos observar la importante elevación de riesgo de cáncer de mama comparado con el 12% de riesgo de nuestra población general76. Se encontró en nuestra serie un porcentaje de presentación de cáncer de páncreas más elevado que el 5% reportado por Korsse et al. (2013) en su trabajo con una cohorte de 144 pacientes holandeses, aunque ellos reportan una edad media de diagnóstico de 55 años, muy similar a la nuestra77. Observamos en las tablas 4 y 5 los elevados RA y RR por edades reportados por este grupo y por otros autores3,29,52,66,67, sobre todo a partir de los 50 años; hay que resaltar que en el trabajo de Korsse para su análisis clasifican y analizan por separado el cáncer de páncreas, el ampular y el de conductos biliares distales, y en los otros trabajos descritos no se especifican las localizaciones anatómicas.
Riesgos acumulados para padecer cáncer en pacientes con SPJ
| Tipo de cáncer | Cualquier cáncer | GI | Esófago | Gástrico | ID | Colorrectal | Páncreas | Pulmón | Test | Mama | Útero | Ovario | Cérvix | Gine | Tiroides | Vejiga | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Edad | % | ||||||||||||||||
| Giardello et al./200029,60,81–83 | 15-64 a | 93 | − | 0.5 | 29 | 13 | 39 | 36 | 15 | 9 | 54 | 9 | 21 | 10 | − | − | − |
| Hearle et al./200666 | 20 a | 2 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 30 a | 5 | 1 | − | − | − | − | − | − | − | − | − | − | − | 1 | − | − | |
| 40 a | 17 | 9 | − | − | − | 3 | 3 | 1 | − | 8 | − | − | − | 3 | − | − | |
| 50 a | 31 | 15 | − | − | − | 5 | 5 | 4 | − | − | − | − | − | 8 | − | − | |
| 60 a | 60 | 33 | − | − | − | 15 | 7 | 13 | − | 31 | − | − | − | 18 | − | − | |
| 70 a | 85 | − | − | − | − | 39 | 11 | 17 | − | − | − | − | − | 18 | − | − | |
| Van Lier et al./20103,29,58,66,74,75 | 60 a | 37-93 | 38-66 | − | − | − | − | − | − | − | 32-54 | − | − | − | 13-18 | − | − |
| 65 a | − | 29 | 13 | 39 | 11-36 | 7-17 | 9 | 9 | 21 | 10 | − | − | |||||
| 70 a | − | − | − | − | − | − | − | − | − | − | |||||||
| Resta et al./201352 | 15-65a | 90 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| 40 a | 20 | 7 | − | − | − | 3 | 2 | − | − | − | − | − | − | − | − | − | |
| 50 a | 43 | 20 | − | − | − | 6 | 4.5 | − | − | − | − | − | 14 | − | − | − | |
| 60 a | 71 | 44 | − | − | − | 12 | 18 | − | − | − | − | − | − | − | − | − | |
| 65 a | 89 | 55 | − | − | − | − | 55 | − | − | − | − | − | 22.7 | − | − | − | |
| Korsse et al./201377 | 50 a | − | − | − | − | − | − | 3.9 | − | − | − | − | − | − | − | − | − |
| 60 a | − | − | − | − | − | − | 11.1 | − | − | − | − | − | − | − | − | − | |
| 70 a | − | − | − | − | − | − | 26 | − | − | − | − | − | − | − | − | − | |
| Ishida et al./201667 | 70 a | − | − | − | 24 | 24.1 | 36.4 | 29.4 M: 36.1 H: 26.6 | 7.6 | − | 19.3 | 46.5 | 10.1 | − | − | 2.7 | 10.5 |
a: años; GI: gastrointestinal (colorrectal, intestino delgado, gástrico, esófago, páncreas); Gine: ginecológico; H: hombre; ID: intestino delgado; M: mujer; Test: testículo.
Por otro lado, llama la atención que los RA para todo tipo de cáncer descritos a los 40 años para un paciente con SPJ son del 17% al 20% y comparados con el informe de la Red Española de Registros de Cáncer 2015, de todos los riesgos informados que tiene la población general de padecer determinado tipo de cáncer según localización de los 0 a los 49 años ninguno supera el 1.86%, que corresponde al riesgo de cáncer de mama en mujeres, por lo que nuestros pacientes a la edad de 40 años superan de forma importante los riesgos de nuestra población general78. Además, las edades medias de diagnóstico de cáncer de todas las series nos hablan que van desde los 36 a 45 años, coincidiendo con la de nuestra serie y resaltando que en nuestro estudio el caso de diagnóstico más temprano de cáncer fue a los 30 años3,13,22,29,39,68. Todo lo anterior nos lleva a pensar que quizá la presentación de cáncer y los RA para presentar cáncer sean mayores en adultos jóvenes y más elevados en los rangos de edad entre los 35 y 50 años, por lo que es de suma importancia el cumplimiento y la continuidad de las medidas de los protocolos descritos para el screening de cáncer hereditario en estos pacientes, y sobre todo antes de llegar a estos rangos de edad.
En concordancia con nosotros se han realizado estudios que realizan una descripción clínica relacionada con la presentación de cáncer; el porcentaje de pacientes que padecen cáncer en nuestra serie es elevado y similar a lo descrito recientemente por Jelsig et al.20 en su estudio danés, que reportan que el 42% de los casos presentaron cáncer en varios sitios anatómicos; también en varias series de casos hablan de que los pacientes que presentaron cáncer fueron del 28% al 67% de todos los casos39,50,68. Es curioso que en el estudio realizado por Tan et al. ninguno presentó cáncer26, quizás puede ser explicado por el número reducido de casos.
El consejo genético en el caso del SPJ brinda muchos beneficios para el paciente con SPJ, su adecuado funcionamiento se debe asegurar si se realiza conjuntamente con un equipo multidisciplinar que implique otras especialidades médicas, laboratorio clínico, enfermería, trabajo social y psicología, pero además va de la mano con las recomendaciones e implementación de protocolos de seguimiento y vigilancia. Se han propuesto múltiples pautas de vigilancia para el cribado de cáncer, la vigilancia de pólipos y manejo integral del SPJ, además hay numerosos protocolos y recomendaciones descritas a nivel mundial; pero, debido a la variabilidad de resultados y las diferentes estimaciones de riesgos, todavía no se ha alcanzado un consenso. En el año 2008 se publicó la Guía de cáncer hereditario en la Comunidad Valenciana y en el año 2017 se realiza una nueva actualización79. El grupo de Tan et al. nos dice que solo 2/7 casos (28%) recibieron consejo genético26; en cambio en nuestra serie la mayoría de los pacientes han acudido a una UCGCH, destacando en este apartado que la primera comunidad autónoma en España en regular el servicio de una UCGCH en cáncer hereditario es la Comunidad Valenciana80.
ConclusionesEl código CIE-9ES 759.6 abarca muchas enfermedades no relacionadas entre sí, lo convierte en poco específico para la búsqueda y registro de pacientes con SPJ. Puede que la clasificación CIE-10E aporte más especificidad, ya que con su uso se podrían localizar 3 ER concretas. Sin embargo, estas enfermedades no tienen ninguna relación, por lo que surge la necesidad de usar otro tipo de clasificación y usar un código individual para cada enfermedad. Es importante unificar y establecer clasificaciones y codificaciones estándar para una ER, y sobre todo debe existir conciencia en lo indispensable, que es hacer un adecuado uso e implementación de estas.
La media de edad diagnóstica de los casos de la provincia de Valencia está dentro de la población pediátrica, por lo que es importante conocer las características y presentación clínica del SPJ en esta población.
El porcentaje de casos que presenta la mutación y el que presentan los casos esporádicos están descritos dentro de un rango muy amplio, apoyando de esta forma la heterogeneidad genética de la enfermedad.
En algunos casos es probable encontrar en estos pacientes pólipos hiperplásicos y adenomatosos; en este último caso, encontrar un pólipo adenomatoso podría ser un signo de alarma.
En los casos de nuestra población los pólipos están localizados con mayor frecuencia en el ID, especialmente en el duodeno.
El SPJ se asocia con un alto riesgo de padecer cáncer a edades tempranas y a una tasa elevada de complicaciones, con mayor frecuencia las invaginaciones intestinales. Se deberían revisar y evaluar los protocolos de vigilancia con el fin de aumentar la expectativa de vida de estos pacientes y disminuir la presentación de complicaciones.
Las pruebas de cribado para el cáncer de páncreas deberían estar incluidas en todos los protocolos de vigilancia del SPJ.
La implementación y el funcionamiento de las UCGCH tienen beneficios y adecuada acogida en los últimos años en el manejo y control de los pacientes con SPJ en nuestra provincia. Sería conveniente que al menos cada país unificara sus protocolos en función de sus características poblacionales y epidemiológicas a las técnicas de laboratorio, pruebas de diagnóstico y tratamientos utilizados. Son necesarios más estudios clínicos y epidemiológicos para determinar características clínicas específicas, riesgos de las complicaciones asociadas y medidas diagnóstico-terapéuticas de los pacientes con SPJ en España, así como también estudios para valorar y medir el impacto sobre la morbimortalidad del consejo genético y los protocolos de vigilancia en la Comunidad Valenciana.
FinanciaciónLa presente investigación no ha recibido ninguna financiación específica de agencias de los sectores público, comercial o sin ánimo de lucro.
Conflicto de interesesLos autores declaran que no existe conflicto de intereses.
Los autores agradecen a todos los pacientes y sus familias por aceptar la participación en el estudio. Agradecen a todas las personas colaboradoras de los departamentos administrativos y de documentación clínica, comités de ética, unidades de consejo genético en cáncer hereditario y departamentos de biología molecular de todos los hospitales colaboradores y del Hospital General Universitario de Elche; al Servicio de Estudios Epidemiológicos y Estadísticas Sanitarias de Enfermedades Raras, Servicio de Promoción de Salud y Prevención en el Entorno del Programa de Cáncer Hereditario de la Subdirección General de Epidemiología, Vigilancia de la Salud y Sanidad Ambiental de la Dirección General de Salud Pública de la Comunidad Valenciana y a Manuel Posada de la Paz.

