
La enfermedad de Wilson se caracteriza por la acumulación de cobre en diversos órganos: hígado, cerebro y córnea. La causa molecular que la provoca son las mutaciones en el gen atp7b. Se han informado en la literatura más de 120 polimorfismos en el gen atp7b. El objetivo del presente trabajo fue identificar los cambios conformacionales en el exón3 del gen atp7b y detectar el polimorfismo p.L456V en pacientes cubanos con diagnóstico clínico presuntivo de la enfermedad de Wilson.
Material y métodosEn el Centro Nacional de Genética Médica y en el Instituto Nacional de Gastroenterología, durante el período 2007-2012 se realizó un estudio descriptivo que incluyó 105 pacientes con diagnóstico clínico presuntivo de la enfermedad de Wilson. La extracción del ADN fue por la técnica de precipitación salina. Se utilizó la técnica de reacción en cadena de la polimerasa para la amplificación del fragmento de interés, y para identificar los cambios conformacionales y la presencia del polimorfismo p.L456V en el exón 3 del gen atp7b se usó la técnica de polimorfismo conformacional de simple cadena.
ResultadosSe identifican los cambios conformacionales denominados b y c, que correspondieron al polimorfismo p.L456V en estado heterocigótico y homocigótico, respectivamente. La frecuencia alélica del polimorfismo p.L456V en 105 pacientes cubanos diagnosticados clínicamente con la enfermedad de Wilson es del 41%. Las manifestaciones más frecuentes en los pacientes que presentaron este polimorfismo son las hepáticas.
ConclusiónSe identificó el polimorfismo p.L456V en 64 pacientes cubanos con diagnóstico clínico de la enfermedad de Wilson, lo cual posibilitará hacer estudios moleculares por métodos indirectos.
Wilson's disease is characterized by the accumulation of copper in different organs, mainly affecting the liver, brain, and cornea, and is caused by mutations in the ATP7B gene. More than 120 polymorphisms in the ATP7B gene have been reported in the medical literature. The aim of the present study was to identify the conformational changes in the exon 3 region of the ATP7B gene and detect the p.L456V polymorphism in Cuban patients clinically diagnosed with Wilson's disease.
Material and methodsA descriptive study was conducted at the Centro Nacional de Genética Médica and the Instituto Nacional de Gastroenterología within the time frame of 2007-2012 and included 105 patients with a clinical diagnosis of Wilson's disease. DNA extraction was performed through the salting-out method and the fragment of interest was amplified using the polymerase chain reaction technique. The conformational shift changes in the exon 3 region and the presence of the p.L456V polymorphism were identified through the Single-Strand Conformation Polymorphism analysis.
ResultsThe so-called b and c conformational shift changes, corresponding to the p.L456V polymorphism in the heterozygous and homozygous states, respectively, were identified. The allelic frequency of the p.L456V polymorphism in the 105 Cuban patients that had a clinical diagnosis of Wilson's disease was 41% and liver-related symptoms were the most frequent in the patients with that polymorphism.
ConclusionThe p.L456V polymorphism was identified in 64 Cuban patients clinically diagnosed with Wilson's disease, making future molecular study through indirect methods possible.
La enfermedad de Wilson (EW; MIM #27790) presenta un patrón de herencia autosómico recesivo. Constituye un problema de salud mundial. Se clasifica como una enfermedad genética rara y tiene tratamiento; si no se atiende, puede provocar daños irreversibles en el hígado y en el cerebro que conducen a la muerte del paciente.
Se caracteriza por la acumulación de cobre en diferentes órganos: hígado, cerebro y córnea. El diagnóstico clínico de esta enfermedad es complejo1. Se caracteriza por daños en el hígado, que pueden manifestarse mediante la elevación de los niveles séricos de transaminasas hasta hepatitis fulminante. Los pacientes con esta enfermedad pueden presentar afectaciones a nivel cerebral. Además, pueden tener trastornos psiquiátricos, como depresión y tendencias suicidas, entre otras.
La causa molecular que provoca la EW son las mutaciones en el gen atp7b, el cual presenta 20 intrones y 21 exones que codifican para la proteína ATP7B, transportadora de cobre en el hepatocito. Se han identificado más de 120 polimorfismos que están distribuidos en todo el gen atp7b y en los intrones. Los exones más polimórficos reportados son: 2, 8 y 162.
El polimorfismo p.L456V se encuentra en el exón 3 del gen atp7b. Este se identifica en diversas poblaciones, tales como China3, Taiwán4, Egipto5 y Venezuela6, entre otras. Este polimorfismo de un solo nucleótido (SNP) se utiliza en la realización de los haplotipos en familias con pacientes con EW y permite el diagnóstico molecular en ellos6.
Para la determinación del espectro de polimorfismos en el gen atp7b se requiere una adecuada tecnología de cribado. Una de las técnicas más utilizadas para este propósito es el polimorfismo conformacional de simple cadena (SSCP). Se basa en la relación que existe entre la movilidad electroforética de la simple cadena de ADN y su estructura tridimensional. Un cambio en la secuencia de ADN provoca un cambio conformacional, que a su vez provoca un cambio de movilidad en la electroforesis detectable por esta técnica, lo que permite identificar en las muestras de ADN cambios conformacionales diferentes a la variante normal7.
ObjetivosIdentificar los cambios conformacionales en el exón 3 y detectar el polimorfismo p.L456V en pacientes cubanos con diagnóstico presuntivo de EW.
Material y métodosEn el Centro Nacional de Genética Médica, durante el período 2007-2012 se realizó un estudio descriptivo que incluyó 105 pacientes (43 mujeres y 62 hombres) con diagnóstico presuntivo de EW, los cuales asistían a las consultas en el Instituto Nacional de Gastroenterología. Estos pacientes dieron su consentimiento para participar en la investigación, de acuerdo con los principios éticos de la declaración de Helsinki.
Las variables analizadas fueron: frecuencia alélica del polimorfismo p.L456V, cambio conformacional a para la variante normal, cambio conformacional b para la presencia del polimorfismo p.L456V en estado heterocigótico, y cambio conformacional c en estado homocigótico. Las manifestaciones clínicas se clasificaron en hepáticas, neurológicas, psiquiátricas y sus combinaciones.
La evaluación de las manifestaciones clínicas se realizó por un equipo multidisciplinario (gastroenterólogos, genetistas, neurólogos y bioquímicos). Se siguieron los criterios establecidos para el diagnóstico de la enfermedad.
Se seleccionó el exón 3 del gen atp7b para la detección de cambios conformacionales y la identificación del polimorfismo p.L456V. A todos los pacientes se les tomó una muestra de sangre y se extrajo el ADN mediante el método de precipitación salina8 a partir de 10ml de sangre periférica con ácido etildiaminotetraacético (EDTA) (56mg/ml). Las condiciones para la amplificación del exón 3 mediante la técnica de reacción en cadena de la polimerasa (PCR), fueron: 100ng de ADN, 10(F) 5’-AGT CGC CAT GTA AGT GAT AA-3’ y (R) 5’-CTG AGG GAA CAT GAA ACA A-3’, 1mM de dNTPs (Boehringer), 10X tampón PCR, 15mM de MgCl2, 0.3U de Taq polimerasa (Invitrogen), en un volumen de 25μl.
Posteriormente se realizó la electroforesis SSCP. Se mezclaron 3.5μl con una solución de parada de bromofenol azul (0.05% BFA, 10mM NaOH, 95% formamida, 20mM EDTA) y 1μl del producto amplificado, en un volumen final de 7μl. Se aplicó en un gel de acrilamida comercial (GeneGel Excel 12,5/24 Kit). La visualización del ADN se realizó por el método de tinción con plata siguiendo las instrucciones del juego comercial kit PlusOne DNA Silver Staining (Amersham Biosciences, 2007).
Los controles (muestras de ADN de pacientes heterocigóticos y homocigóticos para el polimorfismo p.L456V) utilizados fueron donados por el investigador Georgios Loudianos, del laboratorio Ospedale Regionale per le Microcitemie (Cagliari, Italia).
Se analizaron las comparaciones de las frecuencias por la prueba de chi cuadrado.
Esta investigación fue aprobada por el consejo científico y el Comité de Ética del Centro Nacional de Genética Médica y el Instituto Nacional de Gastroenterología.
ResultadosEn los 105 pacientes que presentaron el polimorfismo p.L456V, la edad fue de 31.6±14.1 y el rango es de 11años (mínimo) a 58años (máximo).
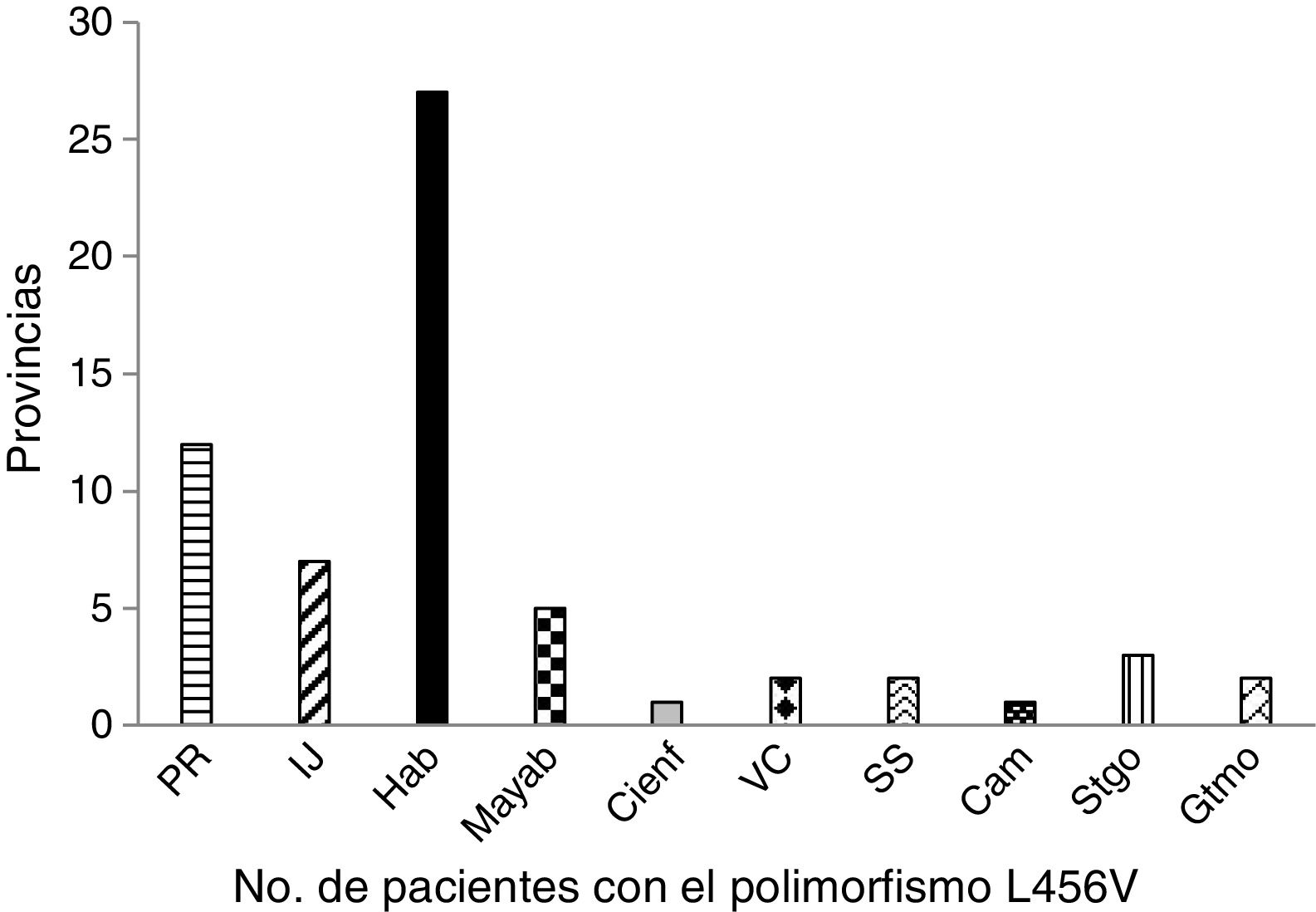
Los pacientes estudiados con el polimorfismo p.L456V estaban representados en 10 provincias y el municipio especial Isla de la Juventud. La provincia más representada es la Habana, seguida de Pinar del Río (fig. 1).

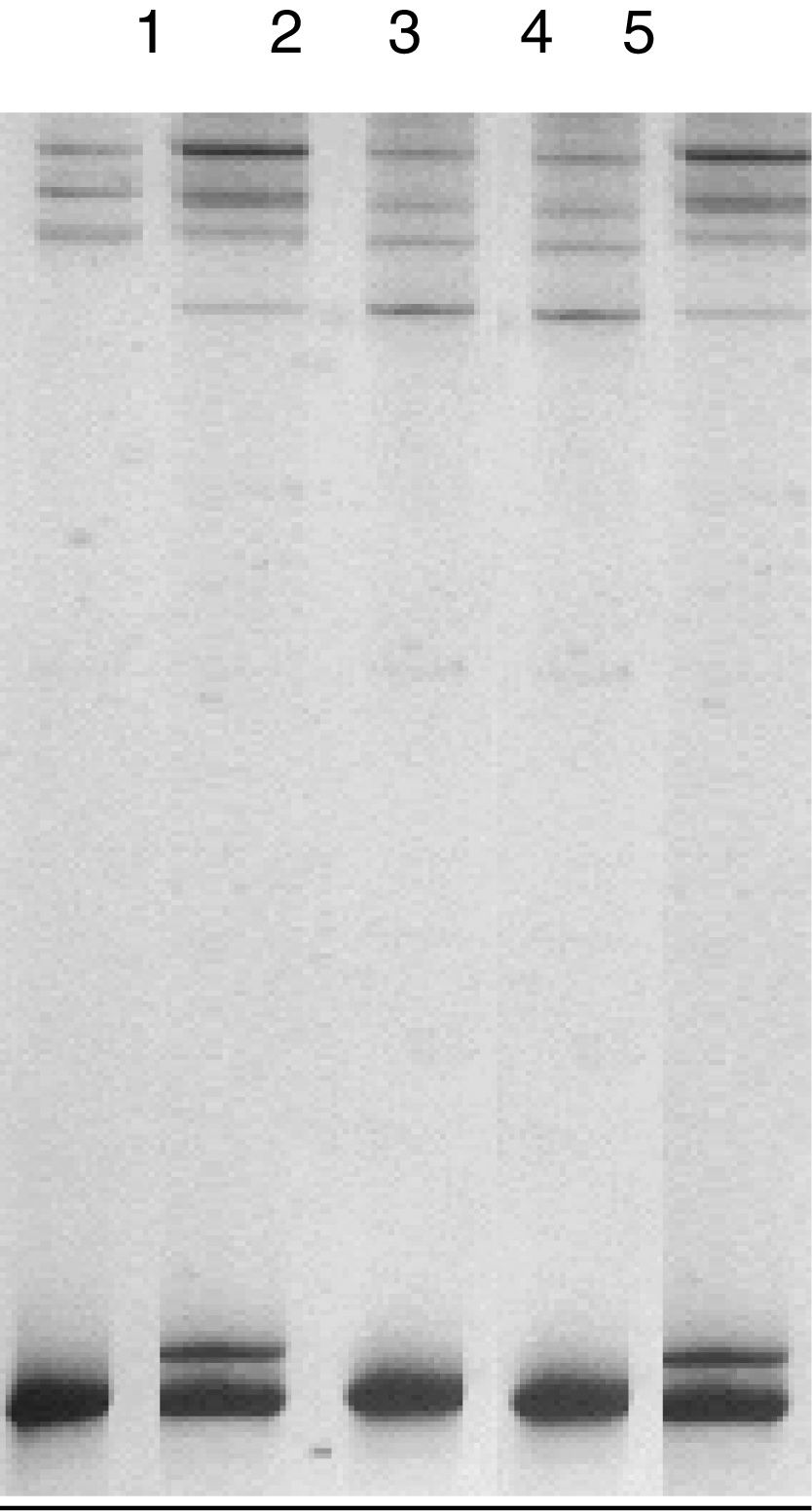
Se identificaron los cambios conformacionales denominados b y c con el uso de la técnica SSCP (fig. 2). El cambio conformacional denominado a correspondió a la variante normal. Los cambios conformacionales b y c correspondieron a la presencia del polimorfismo p.L456V estado heterocigótico y homocigótico, respectivamente.
 del exón 3 del gen atp7b en dos controles positivos para el polimorfismo p.L456V. Visualizado en un gel acrilamida al 12,5%. Las carrileras corresponden a 1: cambio conformacional a, variante normal, control negativo; 2: cambio conformacional b, control heterocigótico para el polimorfismo p.L456V (C-1); 3: cambio conformacional c, control homocigótico para el polimorfismo p.L456V (C-2); 4: cambio conformacional c, homocigótico para el polimorfismo p.L456V; 5: cambio conformacional b, heterocigótico para el polimorfismo p.L456V.")
Polimorfismo conformacional de simple cadena (SSCP) del exón 3 del gen atp7b en dos controles positivos para el polimorfismo p.L456V. Visualizado en un gel acrilamida al 12,5%. Las carrileras corresponden a 1: cambio conformacional a, variante normal, control negativo; 2: cambio conformacional b, control heterocigótico para el polimorfismo p.L456V (C-1); 3: cambio conformacional c, control homocigótico para el polimorfismo p.L456V (C-2); 4: cambio conformacional c, homocigótico para el polimorfismo p.L456V; 5: cambio conformacional b, heterocigótico para el polimorfismo p.L456V.
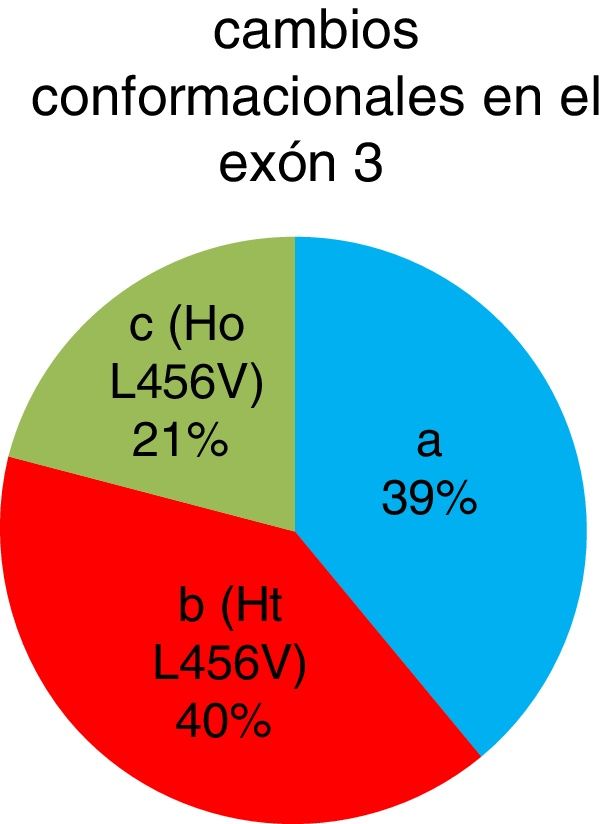
Se detectaron 42 pacientes (40%) que presentaron el cambio conformacional b, 22 pacientes (21%) con el cambio conformacional c y 41 pacientes (39%) con el cambio conformacional a. La presencia del polimorfismo p.L456V fue en un total de 64 pacientes (fig. 3).

De los 105 pacientes cubanos con diagnóstico clínico presuntivo de la enfermedad de Wilson, se identificaron 42 pacientes heterocigóticos y 22 homocigóticos para el polimorfismo p.L456V por la técnica SSCP. La frecuencia alélica del polimorfismo p.L456V en pacientes cubanos estudiados en esta investigación es del 41%.
El polimorfismo p.L456V es consecuencia del cambio de guanina por citocina que provoca un cambio del aminoácido leucina por valina en la posición 456 de la proteína ATP7B. Este cambio no afecta la función de la proteína transportadora de cobre, ATP7B en humanos, y se detectó en diversas poblaciones con una frecuencia mayor del 1%3-5.
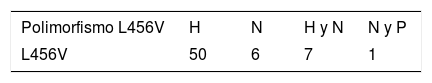
Entre las principales manifestaciones clínicas en los pacientes que presentaron el polimorfismo p.L456V fueron más frecuentes las hepáticas, con el 78%; a continuación las manifestaciones mixtas (hepáticas y neurológicas), con el 11%; las neurológicas, con el 9%, y las mixtas (neurológicas y psiquiátricas), con el 2% (tabla 1).
Principales manifestaciones clínicas presentes en pacientes cubanos que presentan el polimorfismo p.L456V
| Polimorfismo L456V | H | N | H y N | N y P |
|---|---|---|---|---|
| L456V | 50 | 6 | 7 | 1 |
H: manifestaciones hepáticas; N: manifestaciones neurológicas; H y N: manifestaciones hepáticas y neurológicas; N y P: manifestaciones neurológicas y psiquiátricas.
En cuatro pacientes con el polimorfismo p.L456V se identificó la presencia de los anillos de Kayser-Fleischer, lo que representa el 6,3% de los pacientes que se estudiaron, y constituye un criterio de diagnóstico clínico de la enfermedad. Las manifestaciones clínicas de los mismos eran: dos pacientes con manifestaciones hepáticas y neurológicas, un paciente con manifestaciones neurológicas y un paciente con manifestaciones neurológicas y psiquiátricas.
Discusión y conclusionesLa edad de los pacientes con el polimorfismo p.L456V es similar a lo informado en la literatura internacional1.
En la distribución por provincias de los pacientes con el polimorfismo p.L456V se observó que el mayor número de los pacientes eran de la Habana y de Pinar de Río. La mayoría de los pacientes con este polimorfismo se han concentrado en la región Occidental del país.
Se informan más de 120 polimorfismos en el gen atp7b en pacientes con la enfermedad de Wilson2. En Cuba se comienza a realizar la detección del polimorfismo p.L456V en el año 2007, en el Centro Nacional de Genética Médica. Un paso previo a la búsqueda de las mutaciones y los polimorfismos en este gen es la detección de los cambios conformacionales mediante la utilización de la técnica de SSCP. El polimorfismo p.L456V se determinó por comparación de las corridas electroforéticas mediante la técnica de SSCP de controles positivos heterocigóticos y homocigóticos del polimorfismo p.L456V con el producto que se obtuvo de la técnica de PCR del exón3 del gen atp7b de las muestras de los pacientes cubanos que se analizaron.
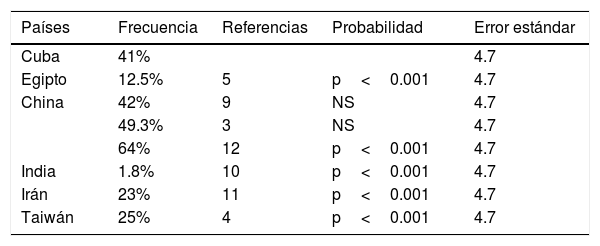
La frecuencia del polimorfismo p.L456V en dos investigaciones en China son similares a nuestro estudio (no hay diferencias estadísticamente significativas), y como se esperaba, mayor del 1%. Hay diferencias estadísticamente significativas al comparar nuestros resultados con los de Egipto, la India, Irán y Taiwán, y la posible explicación es el origen étnico de la población cubana. Como se aprecia en la tabla 2, el resultado que se obtuvo en esta investigación es la segunda mayor frecuencia informada al compararlo con los países que se analizaron. En estudios posteriores se investigará el impacto del polimorfismo p.L456V en los pacientes cubanos con diagnóstico molecular de EW. Es interesante ver que la frecuencia de este polimorfismo es muy baja en la India (1.8%), donde se comporta como una variante rara.
Frecuencias por países del polimorfismo p. L456V en pacientes que presentan diagnóstico clínico de la EW
| Países | Frecuencia | Referencias | Probabilidad | Error estándar |
|---|---|---|---|---|
| Cuba | 41% | 4.7 | ||
| Egipto | 12.5% | 5 | p<0.001 | 4.7 |
| China | 42% | 9 | NS | 4.7 |
| 49.3% | 3 | NS | 4.7 | |
| 64% | 12 | p<0.001 | 4.7 | |
| India | 1.8% | 10 | p<0.001 | 4.7 |
| Irán | 23% | 11 | p<0.001 | 4.7 |
| Taiwán | 25% | 4 | p<0.001 | 4.7 |
Hay investigaciones en varios países que realizaron el estudio molecular en pacientes con EW y no informan la presencia de este polimorfismo9,10. En Turquía, la variante p.L456V no se identificó; sin embargo, se halló en el exón3 (exón donde se localiza el polimorfismo p.L456V) otro polimorfismo, el p.V446L11.
Para el diagnóstico molecular de enfermedades genéticas que posean uno o más genes que contienen un número elevado de exones es necesaria una tecnología de avanzada para lograr el éxito del mismo. Existen diversos estudios moleculares en pacientes con EW12-13. Sin embargo, el médico que atiende al paciente con sospecha de EW siempre debe hacer un minucioso diagnóstico clínico y debe tener en cuenta los criterios que se establecen a nivel mundial14.
En diferentes países se ha identificado el polimorfismo p.L456V, aunque no informan las frecuencias6,15, por lo que no se pueden comparar los resultados obtenidos con esta investigación y en algunos casos no construyen los haplotipos con el uso de diferentes SNP determinados en sus investigaciones y desaprovechan la oportunidad de ampliar el diagnóstico molecular.
Aun cuando se determinan nuevas mutaciones16-19 y se realizan técnicas de microarrays20, en países en vías de desarrollo es necesario que se estudien enfermedades genéticas por biología molecular y que se incorpore la búsqueda de polimorfismos, para prestar el servicio de diagnóstico molecular por métodos indirectos en enfermedades donde presentan un número considerable de exones y pocas mutaciones frecuentes.
En Cuba se trabaja en el establecimiento de la estrategia para el diagnóstico molecular de la EW. Se tiene experiencia por este equipo de trabajo en la identificación de polimorfismos en los exones 1021 y 1322,23 y en el intrón924. Con los polimorfismos que ya se detectaron y con este, en los pacientes cubanos con diagnóstico presuntivo de EW podrán realizarse de haplotipos en las familias cubanas con esta enfermedad, así como la realización del diagnóstico molecular por métodos indirectos. En diferentes países, además de detectar el polimorfismo p.L456V, se informan diversas mutaciones para el establecimiento del diagnóstico molecular de la EW, por ejemplo en China25,26, en la India27 y en Irán28, lo cual posibilita el diagnóstico molecular por métodos indirectos y directos, y realizan sus estrategias en aras de mejorar la calidad de vida de los pacientes con EW.
En relación con el origen étnico de la población cubana, es importante señalar que las Islas del Caribe, incluyendo Cuba, fueron las primeras habitadas por meso-americanos y, más tarde, por amerindios, que algunos investigadores plantean que provenían de Venezuela29,30. Se estima que a la llegada de los españoles existían en la isla más de 100,000 habitantes. En los primeros 50años tras la Conquista, esta población se redujo a unos 5,000 individuos. Los colonizadores españoles extinguieron la población amerindia y comenzaron a trasladar aborígenes desde Norteamérica y Mesoamérica hacia Cuba, y a esclavos desde la costa occidental de África, e introdujeron los negros esclavos del Este de África y del África subsahariana, por lo que la composición genética de la población cubana es resultado, fundamentalmente, de la mezcla de españoles caucásicos y negros africanos31,32. La inmigración española se mantuvo constante desde finales del sigloxv hasta la primera mitad del sigloxx. La historia del mestizaje étnico que por casi cinco siglos tuvo lugar entre esos tres grupos originarios (nativos americanos, europeos y africanos) ha creado la estructura genética de la población cubana actual. La contribución promedio de genes de origen europeo, africano y nativo americano en los individuos estudiados fue del 72, del 20 y del 8%, respectivamente32. Se observó que en la distribución de las mutaciones en el gen atp7b responsables de la EW tiene un papel importante la etnicidad33. En un futuro sería útil estudiar las mutaciones y polimorfismos de este gen en el continente americano y evaluar el papel de la etnicidad en la presencia de los mismos y en la susceptibilidad a la enfermedad.
En conclusión, se dispone de una herramienta molecular para la introducción del diagnóstico molecular en pacientes con EW en Cuba. La metodología puede ser utilizada en otras poblaciones con un perfil genético similar al de Cuba.
Responsabilidades éticasProtección de las personas y los animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoNo se recibió apoyo financiero en relación con este artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses potencial relevante para el artículo de ninguna índole.
A los pacientes por participar en esta investigación, a Lídice Reyes por realizar la extracción de ADN, al Ministerio de Salud Pública, al Dr. Carlos Maragoto por sus aportes, a Georgios Loudianos por el envío de controles positivos, al Dr. Carlos Castañeda por sus aportes en el diagnóstico clínico de la enfermedad.
Este estudio es el resultado del trabajo conjunto del Centro Nacional de Genética Médica (centro colaborador de la OMS para el desarrollo de enfoques genéticos en la promoción de salud), el Instituto Nacional de Gastroenterología y la Universidad de Guantánamo.

