
Los medicamentos biotecnológicos biocomparables son definidos como tratamientos no innovadores que han demostrado calidad, eficacia y seguridad comparable al medicamento de referencia. Los estudios clínicos aleatorizados han demostrado que el biocomparable CT-P13 (infliximab) y los candidatos a biocomparables ABP 501 y ZRC 3197 (adalimumab) no difieren significativamente en la eficacia y seguridad respecto al medicamento innovador en pacientes con otras enfermedades autoinmunes. Sin embargo, se ha generado una controversia sobre el uso de los biocomparables en la enfermedad inflamatoria intestinal ante la incipiente evidencia generada no solo en pacientes sin tratamiento biotecnológico previo sino también en remisión y que podrían ser cambiados al biocomparable por razones no médicas.
Esta revisión es el primer análisis crítico de diversos especialistas en el área de la gastroenterología sobre el uso de biocomparables en enfermedad inflamatoria intestinal, de la evidencia de intercambiabilidad, la extrapolación de indicaciones, eficacia, seguridad, inmunogenicidad y del impacto clínico de la regulación sanitaria en México. El objetivo es compartir el posicionamiento y recomendaciones con respecto a estas nuevas opciones terapéuticas que tienen un potencial de costo-beneficio para el paciente y las instituciones de salud.
The biotechnology-derived medicines known as biosimilars are defined as non-originator treatments that have demonstrated quality, efficacy, and safety comparable to the reference biologic drug. Clinical trials have shown that the infliximab biosimilar, CT-P13, and the candidates for the adalimumab biosimilars, ABP 501 and ZRC 3197, are not significantly different, with respect to efficacy and safety, from the originator drugs in patients with other autoimmune diseases. However, controversy has arisen over the use of biosimilars in inflammatory bowel disease, due to the incipient evidence not only in patients with no previous biotechnology treatment, but also in patients in remission, that could be switched to a biosimilar for non-medical reasons.
The present review is the first critical analysis by different specialists in the area of gastroenterology on the use of biosimilars in inflammatory bowel disease, the evidence on interchangeability, the extrapolation of indications, efficacy, safety, immunogenicity, and the clinical impact of the Mexican health regulations. The aim of our review was to make the positioning and recommendations of these new therapeutic options known, given that they have a potential cost-benefit for both patients and healthcare institutions.
A mediados de los años setenta se realizó un rápido desarrollo de la ingeniería genética que permitió la producción de proteínas recombinantes en cantidades suficientes para utilizarlas terapéuticamente. Este proceso incorpora fragmentos de ADN recombinante, el cual codifica la producción de proteínas específicas dentro de líneas celulares artificiales que han permitido la producción de biofármacos con fines terapéuticos, por ejemplo, los anticuerpos monoclonales1.
La primera generación de medicamentos biotecnológicos ha beneficiado a millones de pacientes en todo el mundo para tratar o prevenir algunas enfermedades, por ejemplo, la enfermedad inflamatoria intestinal (EII), ciertas anemias, algunos tipos de cáncer, enfermedades autoinmunes, deficiencias del crecimiento, problemas de reproducción, diabetes dependiente de insulina y diversas enfermedades con un componente de inflamación crónica2. Con el paso de los años, los medicamentos biotecnológicos han demostrado su eficacia y seguridad en la EII en múltiples estudios clínicos.
En 1998 se aprobó la primera terapia biotecnológica para el tratamiento de la enfermedad de Crohn (EC) y en el 2006 para la colitis ulcerosa crónica idiopática (CUCI). El infliximab (Remicade®) fue el primer anticuerpo monoclonal de tipo quimérico aprobado para estas indicaciones; después lo fue el anticuerpo monoclonal humano adalimumab (Humira®), seguido por el fragmento del anticuerpo monoclonal humanizado certolizumab pegilado (Cimzia®) y, finalmente, el anticuerpo monoclonal humano golimumab (Simponi®) para CUCI. Todos ellos inhiben el factor de necrosis tumoral α (TNFα por sus siglas en inglés) y han demostrado tener un perfil de eficacia y seguridad aceptable en estudios clínicos específicos en EII3.
Tras el vencimiento de la patente de alguno de estos medicamentos innovadores, se ha iniciado un esfuerzo intenso para desarrollar versiones alternativas conocidas como «biocomparables», definidas como los medicamentos no innovadores que han demostrado calidad, eficacia y seguridad comparable a las del medicamento innovador. Los biocomparables pretenden ser estructuralmente idénticos a los medicamentos de referencia, pero ofreciendo un mejor costo-beneficio para el paciente y la institución de salud4.
Es importante destacar que el término «biocomparable» es sinónimo de «biosimilar», solo que en México la legislación tuvo que modificar su nomenclatura para no confundir al prescriptor y al paciente con una marca de medicamentos en nuestro país.
A diferencia de los medicamentos genéricos, las proteínas recombinantes como los anticuerpos monoclonales son moléculas estructuralmente más complejas, por lo que no hay «equivalencia absoluta» entre el medicamento innovador y el biocomparable: siempre presentarán diferencias, aunque mínimas, que determinan la necesidad de solicitar al fabricante los estudios de biocomparabilidad definidos como aquellos necesarios que demuestren calidad, eficacia y seguridad.
El biofármaco CT-P13 es el primer biocomparable del infliximab aprobado para enfermedades inflamatorias autoinmunes tanto en Europa y en Estados Unidos como, recientemente, en México5. El CT-P13 presentó estudios de calidad, farmacológicos y 2ensayos clínicos aleatorizados denominados estudio PLANETAS6 (espondilitis anquilosante) y estudio PLANETRA7 (artritis reumatoide) como evidencia. En particular, la Food and Drug Administration y la European Medicines Agency también autorizaron su uso en otras indicaciones como psoriasis, artritis psoriásica y EII a través de un recurso regulatorio denominado «extrapolación de indicaciones» que indica la exención de estudios clínicos específicos para estas enfermedades tomando en cuenta el total de la evidencia demostrada por el fabricante durante el proceso de registro8.
En México, el CT-P13 (infliximab), conocido como Remsima®, también ha sido aprobado recientemente para todas las indicaciones terapéuticas por medio de la extrapolación de indicaciones. Por lo cual, Remsima® está autorizado para ser empleado en EII tanto en pacientes naïve como en aquellos en remisión. La controversia sobre esta práctica —la extrapolación y la intercambiabilidad entre medicamentos biotecnológicos— ha ido en aumento en nuestro país y a nivel mundial.
El objetivo de este estudio es revisar la evidencia disponible con respecto a la eficacia y seguridad en la intercambiabilidad/sustitución de medicamentos biotecnológicos innovadores y biocomparables en EII. Con base en esto, compartimos la postura y recomendaciones sobre el uso y discutimos la normativa mexicana a este respecto.
Materiales y métodosSe realizó una revisión técnica sobre la evidencia disponible con respecto al mantenimiento de la eficacia y seguridad de los medicamentos biocomparables en pacientes con EII en remisión.
Previamente, en una convocatoria académica para los autores del manuscrito, se programaron varias sesiones presenciales para desarrollar la investigación. Se definieron las siguientes palabras clave limitadas al idioma inglés: biosimilar ulcerative colitis; biosimilar Crohn's disease; biosimilar inflammatory bowel disease; CT-P13; biosimilar infliximab; biosimilar adalimumab; ABP 501; ZRC 3197; biosimilar interchangeability; biosimilar switch, que fueron buscadas en las bases de datos internacionales PubMed® y Ovid®.
Se eligieron solo las publicaciones relevantes para el objetivo de la investigación con base en lo siguiente: 1) existe evidencia de cambio entre medicamento innovador y biocomparable específicamente en pacientes con EII en remisión; 2) son publicaciones en extenso con metodología definida y 3) se evaluó un número relevante de pacientes (no serie de casos ni comunicaciones breves). Las publicaciones que no cumplieron estos 3 aspectos mencionados no fueron consideradas para la revisión. El tiempo límite de publicación electrónica permitido fue de los últimos 5 años, hasta agosto del 2017.
La evidencia fue presentada en sesiones académicas, sintetizada, discutida y consensuada con 9 de los 10 investigadores. No se realizó un análisis sistemático ni estadístico adicional ni se determinó metodológicamente el nivel de calidad de las publicaciones.
ResultadosSe encontraron registrados un total de 918 artículos en las bases de datos usando las palabras clave. Solo 254 publicaciones fueron relacionadas con medicamentos biocomparables, de las cuales 141 fueron manuscritos específicos de EII. Finalmente, de estos se consideraron 57 publicaciones y solo 9 reunieron los criterios de los investigadores para ser revisados y discutidos.
En el caso de ABP 501 (adalimumab) y ZRC 3197 (adalimumab) no se encontró evidencia disponible sobre la eficacia y seguridad después del cambio de formulación: solo en otras enfermedades autoinmunes diferentes a la EII.
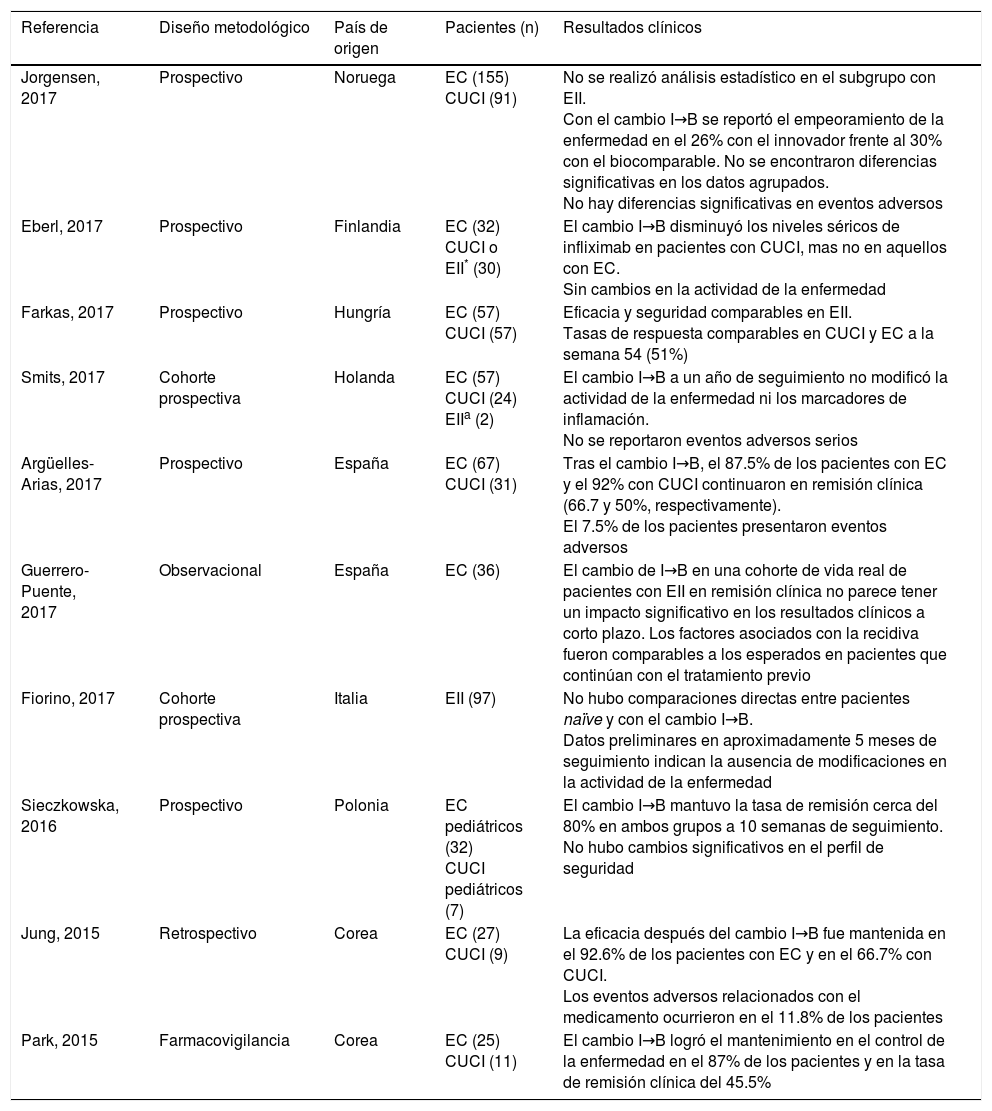
Asimismo, se encontraron 9 estudios de fase IV (farmacovigilancia) que reportan el mantenimiento de la eficacia y seguridad del CT-P13 en pacientes con EII en remisión. Los estudios se muestran en la tabla 1 y son descritos a continuación.
Estudios de intercambiabilidad de CT-P13 en pacientes con diagnóstico de EII
| Referencia | Diseño metodológico | País de origen | Pacientes (n) | Resultados clínicos |
|---|---|---|---|---|
| Jorgensen, 2017 | Prospectivo | Noruega | EC (155) CUCI (91) | No se realizó análisis estadístico en el subgrupo con EII. Con el cambio I→B se reportó el empeoramiento de la enfermedad en el 26% con el innovador frente al 30% con el biocomparable. No se encontraron diferencias significativas en los datos agrupados. No hay diferencias significativas en eventos adversos |
| Eberl, 2017 | Prospectivo | Finlandia | EC (32) CUCI o EII* (30) | El cambio I→B disminuyó los niveles séricos de infliximab en pacientes con CUCI, mas no en aquellos con EC. Sin cambios en la actividad de la enfermedad |
| Farkas, 2017 | Prospectivo | Hungría | EC (57) CUCI (57) | Eficacia y seguridad comparables en EII. Tasas de respuesta comparables en CUCI y EC a la semana 54 (51%) |
| Smits, 2017 | Cohorte prospectiva | Holanda | EC (57) CUCI (24) EIIa (2) | El cambio I→B a un año de seguimiento no modificó la actividad de la enfermedad ni los marcadores de inflamación. No se reportaron eventos adversos serios |
| Argüelles-Arias, 2017 | Prospectivo | España | EC (67) CUCI (31) | Tras el cambio I→B, el 87.5% de los pacientes con EC y el 92% con CUCI continuaron en remisión clínica (66.7 y 50%, respectivamente). El 7.5% de los pacientes presentaron eventos adversos |
| Guerrero-Puente, 2017 | Observacional | España | EC (36) | El cambio de I→B en una cohorte de vida real de pacientes con EII en remisión clínica no parece tener un impacto significativo en los resultados clínicos a corto plazo. Los factores asociados con la recidiva fueron comparables a los esperados en pacientes que continúan con el tratamiento previo |
| Fiorino, 2017 | Cohorte prospectiva | Italia | EII (97) | No hubo comparaciones directas entre pacientes naïve y con el cambio I→B. Datos preliminares en aproximadamente 5 meses de seguimiento indican la ausencia de modificaciones en la actividad de la enfermedad |
| Sieczkowska, 2016 | Prospectivo | Polonia | EC pediátricos (32) CUCI pediátricos (7) | El cambio I→B mantuvo la tasa de remisión cerca del 80% en ambos grupos a 10 semanas de seguimiento. No hubo cambios significativos en el perfil de seguridad |
| Jung, 2015 | Retrospectivo | Corea | EC (27) CUCI (9) | La eficacia después del cambio I→B fue mantenida en el 92.6% de los pacientes con EC y en el 66.7% con CUCI. Los eventos adversos relacionados con el medicamento ocurrieron en el 11.8% de los pacientes |
| Park, 2015 | Farmacovigilancia | Corea | EC (25) CUCI (11) | El cambio I→B logró el mantenimiento en el control de la enfermedad en el 87% de los pacientes y en la tasa de remisión clínica del 45.5% |
I→B: del medicamento innovador al medicamento biocomparable; n: número de pacientes.
La no inferioridad del tratamiento después del cambio entre infliximab de Remicade® y el CT-P13 de Remsima® en sus diferentes indicaciones terapéuticas fue reportada por Jorgensen et al. en el estudio NOR-SWITCH (fase IV): un ensayo aleatorizado (razón 1:1), doble ciego, con 52 semanas de seguimiento. El objetivo primario fue valorar el empeoramiento de la enfermedad después del cambio de tratamiento al biocomparable con un margen de no inferioridad de hasta un 15%, asumiendo que no más del 30% de todos los pacientes empeoraría durante el seguimiento. Se incluyeron 408 pacientes con enfermedad inflamatoria crónica (EC, CUCI, artritis reumatoide, artritis psoriásica, espondilitis anquilosante y psoriasis en placa), que fueron evaluados mediante escalas clínicas respectivas en cada indicación. No se realizaron análisis estadísticos por indicación terapéutica en este estudio.
Los resultados globales indican que 53 de 202 pacientes (26%) con EII empeoraron con el tratamiento innovador (grupo sin cambiar al biocomparable) mientras que 61 de 206 pacientes (30%) empeoraron con el CT-P13 (grupo cambiado al biocomparable). No hubo diferencias significativas en el análisis estadístico global ni en la frecuencia de eventos adversos serios (cerca del 10%) y de inmunogenicidad9.
Por otro lado, Eberl et al. reportaron la ausencia de diferencias significativas en el porcentaje de anticuerpos antiinfliximab después del cambio de terapia. Sin embargo, un mayor número de pacientes con CUCI presentaron niveles séricos de infliximab significativamente menores después del cambio. Este fenómeno farmacológico no fue observado en pacientes con EC10.
Asimismo, la experiencia clínica con CT-P13 fue reportada por Farkas et al. en un estudio de 54 semanas de seguimiento. Pacientes con EC y CUCI fueron monitoreados para evaluar el mantenimiento de la remisión después del cambio de terapia. La tasa de respuesta clínica (51%) se mantuvo comparable en ambos grupos con aquellas alcanzadas a la semana 14 (65.5 y 75.5%, respectivamente)11.
Smits et al. reportaron la ausencia de cambios significativos en la escala de actividad de la enfermedad y en los marcadores inflamatorios en EII durante un año de seguimiento en 83 pacientes monitoreados después del intercambio y solo el 7% de los pacientes interrumpieron la terapia con CT-P13 debido a los eventos adversos. Adicionalmente, se detectó que el 8% de los pacientes fueron positivos a anticuerpos antiinfliximab12.
Resultados semejantes fueron publicados por Jung et al. en un estudio retrospectivo y multicéntrico con 54 semanas de seguimiento en pacientes con EC (n = 27) y CUCI (n = 9). Aquí, el 92.5% de los pacientes mantuvieron la tasa de respuesta clínica alcanzada con el tratamiento previo13.
Recientemente, Argüelles-Arias et al. demostraron la efectividad y seguridad del cambio a CT-P13 en 98 pacientes con EII a los 6 meses de seguimiento. Tanto en EC como en CUCI se mantuvo la remisión alcanzada con el tratamiento previo (66.7 y 50%, respectivamente). No hubo diferencias significativas en el perfil de seguridad después del cambio14.
En el estudio poscomercialización realizado por Park et al., estos reportaron un total del 87% en control de la enfermedad, del cual el 45.5% alcanzó o mantuvo la remisión clínica15.
En el estudio de farmacovigilancia PROSIT-BIO de Fiorino et al. participaron 31 centros hospitalarios en Italia. Los investigadores destacan datos relevantes con respecto al mantenimiento de la eficacia y seguridad en 313 pacientes con EC y en 234 con CUCI, de los cuales 97 pacientes habían sido cambiados al CT-P13. No se encontraron modificaciones en la actividad de la enfermedad a los 5 meses de seguimiento16.
En otro estudio observacional, Guerrero-Puente et al. evaluaron la eficacia, seguridad, biodisponibilidad y los factores asociados con la recaída después del cambio al biocomparable. Se seleccionó a 36 pacientes con EC, no se encontraron diferencias significativas en la respuesta clínica a corto plazo y no hubo modificaciones en la concentración sérica del biofármaco17.
Finalmente, en población pediátrica se encontró poca información: solo destacó el estudio de Sieczkowska et al., quienes mostraron resultados con respecto a la intercambiabilidad al biocomparable en pacientes pediátricos con EII. Fue un estudio prospectivo de colaboración con 3hospitales: en total, se seleccionó a 32 pacientes con EC y a 7 con CUCI. La edad osciló entre 3 y 15 años, con un promedio de 12 años. El mantenimiento de la remisión se conservó en el 80% de los pacientes, sin modificaciones significativas después del cambio de tratamiento a las 10 semanas de monitoreo. El perfil de seguridad del CT-P13 fue consistente con el reportado por el medicamento de referencia18.
DiscusiónEl objetivo de esta investigación fue revisar la evidencia disponible con respecto al mantenimiento de la eficacia y seguridad de los medicamentos biocomparables en pacientes con EII en remisión. Previamente, se han descrito los estudios de eficacia y seguridad en pacientes naïve19. Sin embargo, existe gran controversia sobre la evidencia en el mantenimiento después del cambio del medicamento biotecnológico innovador al biocomparable, así como sobre el vacío en la regulación sanitaria con respecto a la intercambiabilidad entre medicamentos biotecnológicos. En nuestro mejor conocimiento, no existe una publicación previa que aborde este tema en nuestro país.
Debido a que los candidatos a biocomparables de adalimumab ABP 501 y ZRC 3197 todavía no se comercializan en México y no se encontraron estudios disponibles, no fue posible describir una discusión sobre su uso.
En general, la experiencia clínica con el biocomparable Remsima® CT-P13 (infliximab) tiende a resultados positivos que apoyan el uso en EII. De acuerdo a los resultados, el cambio de la terapia, al parecer, no modifica los marcadores inflamatorios de la enfermedad y tampoco impacta negativamente en el mantenimiento de la remisión lograda con el tratamiento previo. No obstante, habrá que considerar que en México aún no existe experiencia clínica suficiente debido, al menos en parte, a la reciente aprobación de Remsima®, por lo que es necesario generar experiencia y determinar la carga económica y social que implican estas enfermedades tanto para el paciente como para el sistema de salud.
Observamos que la información disponible es poca y que sus criterios metodológicos son heterogéneos, lo que, en parte, es comprensible, al tratarse de estudios en fase IV (farmacovigilancia) y ante la falta de ensayos iniciales del CT-P13 en EII. Por lo tanto, nuestra investigación podría presentar la limitante de no haber determinado el nivel de la evidencia mediante un análisis sistemático o estadístico adicional (por ejemplo, un metaanálisis) debido a que nuestra investigación es una revisión bibliográfica que aproximará al especialista al tema de los biocomparables. Consideramos que evaluaciones posteriores serían necesarias para profundizar en la evidencia.
Sin embargo, las implicaciones médicas que ocasionaría la introducción de biocomparables en la EII las describimos a continuación.
Marco legal en MéxicoEn el artículo 222 bis del Reglamento de Insumos de la Ley General de Salud se define un medicamento biocomparable como aquel medicamento no innovador que ha demostrado tener calidad, eficacia y seguridad comparables con el medicamento de referencia20. Además, los requisitos técnicos de biocomparabilidad se encuentran descritos en las Normas Oficiales Mexicanas 257, 177 y 220, en las cuales se basa la autoridad sanitaria, al menos en parte, para emitir el registro sanitario de un medicamento biocomparable21-23.
No encontramos definido en un documento o guía oficial los términos y criterios específicos para la intercambiabilidad y extrapolación entre medicamentos biotecnológicos lo cual, potencialmente, provocaría incertidumbre sobre su práctica.
IntercambiabilidadLa intercambiabilidad se conoce como la sustitución de un medicamento por otro de la misma composición farmacéutica, que se espera que tenga el mismo efecto terapéutico y que mantenga la eficacia y seguridad alcanzadas en pacientes estabilizados con el tratamiento previo24.
La intercambiabilidad entre medicamentos biotecnológicos es un tema controversial. En los Estados Unidos, la Food and Drug Administration ha manifestado oficialmente que la intercambiabilidad compete a cada estado, y permanece neutral en el tema. Por otro lado, la European Medicines Agency tampoco indica recomendaciones ni requiere estudios de intercambiabilidad para el registro sanitario de biocomparables, dejando la decisión a cada país miembro, al médico o al farmacéutico25.
En la normativa mexicana no encontramos una clara definición del proceso para demostrar intercambiabilidad entre innovador y biocomparable, a diferencia de la definición de medicamentos genéricos, en la que el Consejo de Salubridad General en México a través de la comisión de expertos para las pruebas de intercambiabilidad determina una serie de requisitos metodológicos para demostrar esta cualidad entre medicamentos con la misma composición cuali-cuantitativa22. Sin embargo, la regulación sanitaria de medicamentos genéricos no es aplicable a los biocomparables por las claras diferencias farmacológicas entre ellos y por los diferentes procesos de producción1,2.
Además, la adquisición del medicamento biotecnológico, en la mayoría de las ocasiones, es por medio de una institución pública debido su alto costo y al control en la farmacia. En este escenario, tanto el medicamento innovador como el biocomparable legalmente comparten la misma clave del sector salud20, no se hace distinción entre ellos, hecho por el cual una institución de salud puede promover el cambio de la terapia sin consentimiento del paciente y del médico o con evidencia que demuestre su eficacia. Esto deja un vacío importante en la trazabilidad real del tratamiento y la eficiencia en la operatividad de la farmacovigilancia. No encontramos motivo válido para cambiar la terapia biotecnológica de un paciente que se encuentra en remisión gracias a un tratamiento previo, independientemente del costo del tratamiento. Consideramos que esta práctica no médica contrapone los principios bioéticos de la medicina y el derecho de protección a la salud.
Sin embargo, reconocemos que las ventajas de compartir la clave del sector salud recaen en mayor competitividad de precios en este tipo de insumos que potencialmente se traduciría en mayor acceso al paciente y en la minimización en el desabasto de medicamentos. Por lo tanto, el cambio del tratamiento innovador al biocomparable es debatible.
La Food and Drug Administration publicó recientemente una guía general para evaluar la intercambiabilidad de productos biotecnológicos. En ella, recomienda realizar estudios prospectivos que evalúen la no inferioridad del tratamiento en al menos 2 cambios de formulación26.
El estudio de fase IV diseñado específicamente para demostrar la intercambiabilidad entre infliximab, el medicamento innovador, frente al CT-P13 en todas las indicaciones terapéuticas fue el estudio NOR-SWITCH, un ensayo independiente sin financiamiento del fabricante. Aunque los autores no realizaron comparaciones por subgrupos, nosotros analizamos los datos de manera puntual y encontramos que 14 de 78 pacientes (17.9%) con EC empeoraron con el tratamiento innovador (grupo no cambiado al biocomparable) en contraste con 23 de 77 pacientes (29.8%) con CT-P13 (grupo cambiado al biocomparable) en un tiempo de seguimiento de 54 semanas. A su vez, 3 de 47 pacientes (6.3%) con CUCI empeoraron con el tratamiento innovador (grupo no cambiado) mientras que 5 de 46 pacientes (10.8%) empeoraron con CT-P13 (grupo cambiado al biocomparable). Más subanálisis estadísticos aportarían evidencia sobre la consistencia de las conclusiones del estudio.
Los resultados NOR-SWITCH9 generaron controversias durante la elaboración de este manuscrito. Por ejemplo, 1) en la aleatorización de los pacientes no se consideró el fenotipo de la enfermedad o la definición de «estable», lo cual se refleja en un mayor sesgo en la conclusión de los resultados; 2) no se definió qué pacientes estaban en remisión endoscópica; 3) los resultados estadísticos de no inferioridad mostraron una alta variabilidad en la respuesta clínica en cada subgrupo poblacional, no así en el conjunto total; 4) cabe destacar que en EII la respuesta fue menos variable comparada con las de las otras poblaciones; no obstante, resulta controversial la decisión de tomar como objetivo primario el empeoramiento de la enfermedad, ya que la no inferioridad se refiere a un término estadístico y el empeoramiento, a un término clínico; 5) las escalas utilizadas para valorar la actividad de la enfermedad son clínicas, sin tomar en cuenta las escalas endoscópicasy 6) no se presenta estratificación por subgrupos de los datos de seguridad después del cambio.
Un estudio sólido que considerase estas variables aportaría un mayor nivel de evidencia y solidez para demostrar la intercambiabilidad entre infliximab innovador y biocomparable. Los resultados de NOR-SWITCH no son aún concluyentes para demostrar intercambiabilidad y es conveniente esperar los resultados en su fase de extensión.
InmunogenicidadEl proceso de inmunogenicidad, es decir, la producción de anticuerpos antifármaco, es de relevancia clínica debido a que tienen el potencial de modificar la respuesta terapéutica o ciertos aspectos farmacocinéticos-farmacodinámicos. Por ahora, contamos con pruebas especiales de laboratorio para medir el nivel de anticuerpos antifármaco (por ejemplo, prueba de ELISA). Lamentablemente, es poco frecuente su medición rutinaria en la práctica clínica debido a la falta de esta tecnología en nuestros hospitales, además de quela heterogeneidad en la estandarización de la metodología analítica por parte de los laboratorios, la baja sensibilidad en presencia del biofármaco y la gran variabilidad de resultados reportados en la literatura son factores que nos limitan el uso frecuente.
Se ha demostrado que la inmunogenicidad depende en gran parte del grado de «humanización» genética de la proteína recombinante27, independientemente de si es innovadora o biocomparable, mientras los biofármacos sean semejantes en su estructura y conserven la calidad del medicamento de manera consistente entre lote y lote28.
En teoría, los biocomparables no deberían producir mayor inmunogenicidad comparados con el innovador, debido a que conservan los mismos epítopos inmunodominantes, que son regiones de aminoácidos que actúan como determinantes antigénicos con la capacidad de producir anticuerpos. Ben-Horin et al. demostraron que los anticuerpos antiinfliximab sistémicos (policlonales) que circulan en el paciente no son diferentes y reconocen a la misma molécula, el infliximab29.
También Baert et al. reportaron que la presencia sistémica de anticuerpos antiinfliximab aumenta el riesgo de reacciones adversas durante la infusión y se relaciona con la pérdida de la respuesta en pacientes con EII30. Del mismo modo, Farrel et al. observaron que pacientes en remisión resultaron con menor concentración sérica de anticuerpos antiinfliximab, de manera similar a los inmunosuprimidos con premedicación, al contrario de aquellos que perdieron la respuesta clínica31.
En pacientes sensibilizados previamente con alguna terapia biotecnológica existe poca información. Bálint et al. hallaron que estos desarrollaron mayor frecuencia de reacciones adversas periinfusión y presentaron un aumento significativo en la concentración sérica de anticuerpos antiinfliximab comparados con pacientes naïve32. Estudios recientes han confirmado los mismos hallazgos y han sido descritos ampliamente por Hindryckx et al.33.
Con base en lo anterior, proponemos que, en el caso excepcional de encontrar aumento en la inmunogenicidad con el intercambio de la terapia, debe existir algún mecanismo fisiopatológico, aún no entendido completamente, por el cual el sistema inmunológico «rompe su tolerancia» al medicamento biocomparable y, por ende, el cambio al biocomparable podría ser un factor de riesgo. Recomendamos, de ser posible, la medición de anticuerpos antifármaco antes y después del intercambio en un tiempo razonable con el propósito de descartar variables que afectarían la remisión clínica del tratamiento en pacientes previamente sensibilizados, además de tomar en cuenta las otras causas conocidas, tales como, la premedicación/comedicación inmunosupresora, la concomitancia de medicamentos no biotecnológicos, el esquema de optimización del tratamiento, el perfil de glicosilación heterogéneo del biocomparable con respecto al innovador y las posibles inconsistencias en la calidad de la fabricación del medicamento, entre otros factores.
Por otro lado, el tema de las infecciones oportunistas que aparecen por la inhibición del TNFα en el tratamiento a largo plazo también debe abordarse integralmente en los ensayos clínicos, debido a que han sido reportados problemas durante la infusión intravenosa o los derivados del uso del dispositivo médico para aplicar la dosis subcutánea34. Sin embargo, los estudios del biocomparable abundan poco en este tema.
Finalmente, el perfil de seguridad a largo plazo puede ser un diferenciador clave al momento de la intercambiabilidad entre medicamentos biotecnológicos. Por ejemplo, en el estudio PLANETAS (espondilitis anquilosante) en su fase de extensión a 102 semanas, se reportó que el tratamiento no fue inferior después del cambio al CT-P13, al igual que en la fase de extensión en el estudio PLANETRA (artritis reumatoide)35,36. Cabe destacar que la frecuencia de eventos adversos en pacientes con espondilitis anquilosante cambiados al biocomparable fue de 71.4% mientras que en el grupo de pacientes naïve iniciados con CT-P13 fue de 48.9%. Se observa una diferencia del 22.5% entre grupos con el mismo CT-P13 y esto podría apoyar nuestra recomendación de generar evidencia específica en EII debido a que las variables clínicas en remisión y naïve son diferentes, no necesariamente extrapolables.
Extrapolación de indicaciones en enfermedad inflamatoria intestinalSe ha definido como extrapolación de indicaciones el recurso regulatorio en el que se exenta de estudios clínicos al medicamento biotecnológico en determinadas indicaciones terapéuticas con el propósito de obtener un permiso de comercialización, tomando en cuenta el total de la evidencia demostrada por el fabricante durante el proceso de registro, en términos de calidad, eficacia y seguridad en las indicaciones principales8.
En México, la extrapolación se decide en el ámbito regulatorio después de una evaluación «caso por caso» de los factores relacionados con la estructura bioquímica y la función de un biocomparable con base en la opinión de comités de expertos de la Comisión Federal para la Protección contra Riesgos Sanitarios37.
Los criterios técnico-científicos en que se basan las agencias sanitarias para conceder la extrapolación son: 1) que ambos biofármacos tengan el mismo mecanismo farmacológico; 2) que presenten la misma vía de administración y composición farmacéutica; 3) que la eliminación del biofármaco sea lineal (dosis dependiente); 4) que presenten estudios de bioequivalencia farmacocinética; 5) que presenten estudios clínicos en modelos suficientemente sensibles y 6) que se haya caracterizado de manera sólida el perfil de seguridad, no solo la inmunogenicidad, entre otros aspectos38-41.
Sin embargo, la evidencia ha demostrado que el mecanismo de acción farmacológico y el comportamiento farmacocinético de los anti-TNFα no son lo mismo en todos ellos42, lo cual explicaría, al menos en parte, las diferencias clínicas que encontramos en la práctica médica. Esto nos lleva a reflexionar seriamente sobre el impacto clínico de la extrapolación de indicaciones en EII, debido a que el mecanismo no es estrictamente el mismo y en su momento no fue caracterizado de manera amplia por el fabricante del medicamento innovador.
No obstante, discutimos los potenciales beneficios de la extrapolación y concluimos que recaerían en varios aspectos: 1) el propósito de no exponer al paciente a riesgos innecesarios que podrían presentarse en un estudio redundante, lo cual implica un debate bioético respecto al beneficio de la investigación clínica; 2) la dificultad de realizar un estudio sólido, por el limitado tamaño de la muestra en EII; 3) su financiamiento, lo que aumentaría el costo del tratamiento y 4) estimular la competencia económica para favorecer el acceso a un mayor número de pacientes mediante la aprobación expedita de biocomparables.
Concluimos que la extrapolación induce incertidumbre y se origina debido a que el recurso regulatorio subyace en la evaluación del medicamento y no en las variables clínicas asociadas a la enfermedad, que modifican la respuesta del medicamento de manera importante y no son consideradas en los estudios clínicos, necesarios para apoyar la prescripción. Esto implica una desventaja en la práctica médica, debido a que la toma de decisiones por parte del médico especialista siempre es basada en la evidencia clínica y en la experiencia generada con el medicamento previo.
La aceptación médica generalizada sobre la extrapolación de indicaciones como recurso regulatorio no será tarea fácil para los fabricantes de biocomparables en EII mientras no se procuren los canales de comunicación adecuados más allá de aspectos normativos de autorización de medicamentos, no siempre cercanos al prescriptor y al paciente.
Posicionamiento y recomendaciones- 1.
Apoyamos la introducción de los medicamentos biocomparables en la enfermedad inflamatoria intestinal en nuestro país.
- 2.
Sin embargo, la biocomparabilidad sigue siendo un tema con poca difusión dentro de la comunidad médica especialista de la enfermedad.
- 3.
Aunque no existe un registro epidemiológico sobre EII en México, los centros de referencia nacional han observado una tendencia al alza en el número de casos, lo cual implica un mayor requerimiento de terapias biotecnológicas o de nuevas opciones de tratamiento. Los medicamentos biocomparables son una alternativa terapéutica para lograr un mayor acceso en beneficio de los pacientes con EII.
- 4.
Debido a que la EII tiene variables clínicas diferentes a otras enfermedades inflamatorias autoinmunes, recomendamos realizar estudios clínicos específicos en la indicación. Lo anterior tiene como propósito recomendar el uso de biocomparables con mayor certeza y con la máxima información a los pacientes que acuden a consulta médica.
- 5.
En la actualidad, ningún medicamento biocomparable tiene definida claramente la evidencia específica para ser extrapolable de la enfermedad reumática a EII, por lo tanto, su recomendación no debe ser generalizada. Estaríamos de acuerdo con la extrapolación de indicaciones siempre y cuando la evidencia se haya definido previamente bajo un consenso oficial o exista difusión por parte de las agencias sanitarias o el fabricante del biocomparable sobre los criterios con los que sustentaron el permiso de comercialización.
- 6.
México cuenta con una normativa líder en Latinoamérica para la evaluación de medicamentos biocomparables; no obstante, los criterios para la extrapolación de enfermedades reumáticas a EII son aspectos con poca claridad, por lo tanto, se necesita mayor evidencia para su recomendación.
- 7.
La falta de una definición clara de intercambiabilidad entre biotecnológicos en México es un vacío legal y provoca que el concepto clínico se vea como un tema administrativo, sin tomar en cuenta la evidencia médico-científica necesaria para establecer un pronóstico preciso del paciente.
- 8.
Estamos a favor de la intercambiabilidad entre productos biotecnológicos, siempre y cuando la decisión sea consensuada entre el médico y el paciente, además de ser sustentada con el máximo nivel de evidencia científica.
- 9.
No recomendamos la intercambiabilidad o sustitución automática por indicaciones no médicas debido al riesgo potencial de modificar la respuesta al tratamiento previo y a los dilemas bioéticos que se presentarían en la relación médico-paciente.
- 10.
Recomendamos a los fabricantes de biocomparables que realicen estudios de intercambiabilidad específicos en EII desde el desarrollo clínico inicial.
- 11.
Se invita a la comunidad médica gastroenterológica y especialista en EII, así como a los profesionales de la salud, a reportar todos los eventos adversos y la falta de eficacia de todos los medicamentos para impulsar la cultura de la farmacovigilancia dentro de la práctica clínica.
Ninguno de los autores recibió financiamiento ni pago de ninguna especie para la realización del presente manuscrito.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLa postura expresada por los expertos es de carácter unánime y no refleja necesariamente la visión de la institución en que laboran cada uno.
Los Drs. C.M. del Real, B. Rubio y M. Rojas declaran haber recibido beca académica de Abbvie, Pfizer, Janssen y UCB para asistir al menos a un congreso médico. La Dra. Y. Gutiérrez declara haber recibido honorarios de Takeda por impartir conferencias y una beca académica de Abbvie para la asistencia, al menos, a un congreso médico. La Dra. X. Sánchez declara haber recibido beca académica de Janssen para la asistencia, al menos, a un congreso médico.
El Dr. A. Mayoral declara haber recibido honorarios de Abbvie, Pfizer y Janssen por impartir conferencias y una beca académica de Abbvie, Pfizer y UCB para la asistencia a congresos médicos así como por servicios de consultoría de marca por parte de Janssen. El Dr. A. Esquivel declara haber recibido honorarios profesionales de Abbvie y Pfizer por impartir conferencias y por consultoría de marca. El Dr. J.L. Rocha declara haber recibido beca de Abbvie, Pfizer, Janssen y UCB para la asistencia, al menos, a un congreso médico y honorarios profesionales de Abbvie, Pfizer y Janssen por impartir conferencias. Finalmente, el Dr. J. Yamamoto declara haber recibido honorarios de Abbvie, Pfizer, Janssen y UCB por impartir conferencias y una beca académica de Abbvie para la asistencia, al menos, a un congreso médico así como honorarios profesionales por parte de Bristol por haber participado en un estudio clínico. El Dr. J. Ramos declara no tener conflictos de intereses.
Los autores agradecen la valiosa participación de la Dra. Azucena Casanova y del Dr. Alejandro García Martínez para la elaboración del manuscrito.

