

Lysosomal acid lipase deficiency (LAL-D) causes progressive cholesteryl ester and triglyceride accumulation in the lysosomes of hepatocytes and monocyte-macrophage system cells, resulting in a systemic disease with various manifestations that may go unnoticed. It is indispensable to recognize the deficiency, which can present in patients at any age, so that specific treatment can be given. The aim of the present review was to offer a guide for physicians in understanding the fundamental diagnostic aspects of LAL-D, to successfully aid in its identification.
MethodsThe review was designed by a group of Mexican experts and is presented as an orienting algorithm for the pediatrician, internist, gastroenterologist, endocrinologist, geneticist, pathologist, radiologist, and other specialists that could come across this disease in their patients. An up-to-date review of the literature in relation to the clinical manifestations of LAL-D and its diagnosis was performed. The statements were formulated based on said review and were then voted upon. The structured quantitative method employed for reaching consensus was the nominal group technique.
ResultsA practical algorithm of the diagnostic process in LAL-D patients was proposed, based on clinical and laboratory data indicative of the disease and in accordance with the consensus established for each recommendation.
ConclusionThe algorithm provides a sequence of clinical actions from different studies for optimizing the diagnostic process of patients suspected of having LAL-D.
La deficiencia de lipasa ácida lisosomal (DLAL) ocasiona el almacenamiento de ésteres de colesterol y triglicéridos en los lisosomas de los hepatocitos y células del sistema monocito-macrófago y, como consecuencia, produce una enfermedad sistémica con manifestaciones variadas que puede pasar inadvertida; es indispensable reconocerla ya que puede diagnosticarse a cualquier edad y recibir tratamiento específico. El objetivo de este documento es ofrecer una guía que permita al médico conocer los aspectos fundamentales relacionados con el diagnóstico de la DLAL para garantizar su identificación. Este documento fue diseñado por un grupo de expertos y se presenta como un algoritmo para orientar al médico pediatra, internista, gastroenterólogo, endocrinólogo, genetista, patólogo, imagenólogo y otros especialistas que pudieran enfrentar a esta entidad.
MétodosSe realizó una revisión actualizada de la literatura con respecto a las manifestaciones clínicas y el diagnóstico de la DLAL por parte de los expertos mexicanos. Se plantearon las declaraciones con base en esta revisión y se sometieron a votación. Se utilizó el método cuantitativo estructurado de técnica de grupo nominal para alcanzar un consenso.
ResultadoSe propone un algoritmo práctico del proceso diagnóstico de pacientes con DLAL, con base en datos clínicos y de laboratorio indicativos de la enfermedad, acorde con el consenso estabilizador para cada recomendación.
ConclusiónEste algoritmo proporciona una secuencia de acciones clínicas, basado en las manifestaciones clínicas obtenidas de los diferentes estudios, con el propósito de optimizar el proceso diagnóstico de los pacientes con sospecha de DLAL.
Lysosomal acid lipase deficiency (LAL-D) is a recessive autosomal disease characterized by the progressive accumulation of cholesteryl esters and triglycerides in the lysosomes of hepatocytes and the monocyte-macrophage system. It is not exclusive to children and has been diagnosed in persons of all ages. It is a poorly-recognized cause of dyslipidemia, associated with the development of atherosclerosis, cardiovascular disease, and progressive liver disease. It typically presents as hepatomegaly, elevated aminotransferases, and diffuse microvesicular steatosis in liver biopsy.1,2
LAL-D is a disease that can go unnoticed if not suspected, or confused with other entities, such as nonalcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis, heterozygote familial hypercholesterolemia, or familial combined hyperlipidemia, among others.2
The aim of the present work was to propose a quick guide for the suspicion and identification of LAL-D through a proposed diagnostic algorithm.
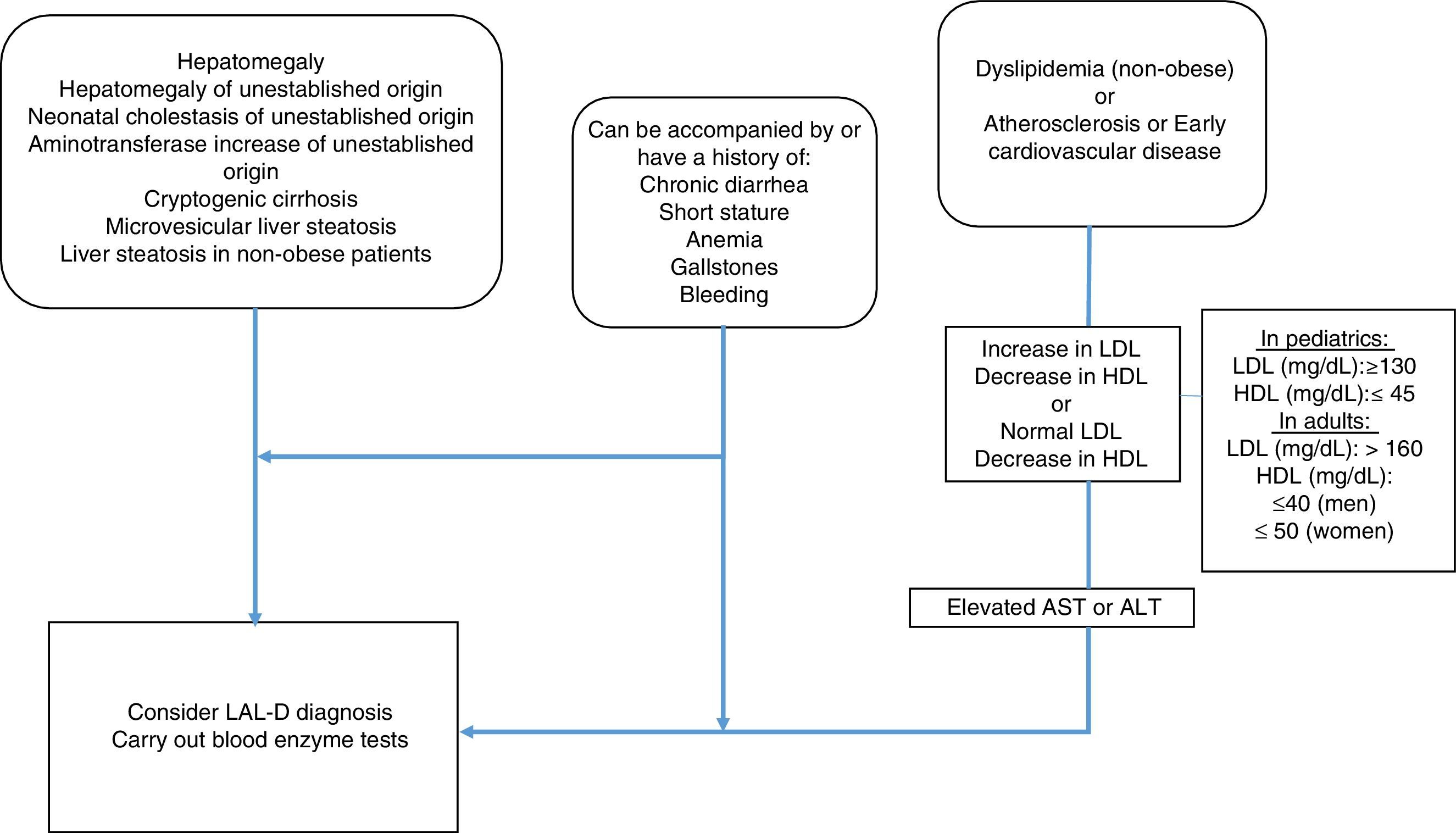
MethodologyMexican experts carried out a systematized review of the literature in relation to the clinical manifestations and diagnosis of LAL-D. Fifteen specialists in the field, with knowledge of LAL-D, were summoned to develop the consensus. The information search was conducted utilizing the following search algorithm in the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE (PubMed), EMBASE (Ovid), and LILACS databases: (“lysosomal acid lipase deficiency” [Mesh] OR “wolman disease” [Mesh] OR “cholesterol ester storage disease” [Mesh]) AND (“2000/01/01” [PDAT]: “2016/02/28” [PDAT]) AND “humans” [MeSH Terms] AND (English[lang] OR Spanish[lang]) AND (“infant”[MeSH Terms] OR “child”[MeSH Terms] OR “adolescent” [MeSH Terms] OR “adult” [MeSH Terms]). Only articles in Spanish and English were evaluated that mentioned clinical manifestations and laboratory and pathology results of LAL-D. The articles were analyzed by the following 5 groups of consensus participants: 1) gastroenterologists (for adults and children), 2) geneticists and internists, 3) endocrinologists (for adults and children), 4) radiologists, and 5) pathologists. The statements were formulated by the groups and then voted upon. The structured quantitative method employed for reaching consensus was the nominal group technique. Voting was done in person, but anonymously, using an electronic device. The statements that reached a consensus value greater than 90% were included and the diagnostic algorithm based on the resulting consensus was constructed (fig. 1).
Results
The systematic search produced 373 articles, none of which corresponded to clinical practice guidelines or clinical trials. Repeated articles, letters to the editor, or case reports contemplated in other articles were eliminated, leaving a total of 184 articles reporting on clinical manifestations and laboratory and imaging data related to LAL-D that were then reviewed by the consensus participants.
DefinitionLAL-D is a lysosomal storage disease that is autosomal recessive and is caused by mutations of the lysosomal acid lipase (LIPA) gene. The reduced enzymatic activity manifests as a progressive accumulation of cholesteryl esters and triglycerides in the liver, spleen, and other organs.1 It can present as progressive liver disease, early atherosclerosis, or gastrointestinal alterations.2
The degree of lysosomal acid lipase (LAL) enzyme deficiency is variable, but does not determine the heterogeneity of the clinical symptoms. There are two phenotypes: that of early presentation, known as Wolman disease, which is severe and progresses rapidly, and without treatment causes death before the patient reaches one year of age; and that of late presentation, or cholesterol ester storage disease (CESD), which is less severe and can manifest at any age, with only 19% of cases presenting in patients below two years of age and the rest in older children and adults.3–6
All children with disease presentation in infancy (Wolman disease) that do not receive treatment, die within the first year of life.
EpidemiologyThe frequency of LAL-D in different populations is unknown, perhaps due to its under-diagnosis.7 Incidence varies from 1 in 40,000 in Germany8,9 to 1 in 700,000 in other populations, such as that of Australia.2,10 Based on those estimates, there would be 750 to 1,500 patients with LAL-D in the United States, alone.10,11
Population studies that evaluated the most frequent CESD mutation produced a carrier frequency of 1 in 1,000 in Asian populations and 1 in 300 in Caucasian and Hispanic populations, supporting the hypothesis that the disease is under-diagnosed.
LAL-D is frequently unnoticed.
PathophysiologyLAL is essential for lipid metabolism. It hydrolyzes cholesteryl esters and triglycerides in low-density lipoprotein (LDL) particles and produces cholesterol and free fatty acids that, in turn, participate in the regulation of cholesterol homeostasis.2,12 Under normal conditions, an excess of intracellular free cholesterol produces: a) reduced LDL-receptor activity, b) inhibition of the enzymatic activity of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through a negative feedback mechanism, reducing endogenous cholesterol synthesis, and c) acyl-coenzyme A:cholesterol acyltransferase stimulation that increases cholesterol esterification.13
When LAL enzyme activity is reduced or absent, cholesteryl esters and triglycerides accumulate in the liposomes, producing intracellular free cholesterol depletion, a reduced capacity for suppressing 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity, and increased endogenous production of cholesterol.14 That increase inhibits LDL-receptor activity, causing a reduction in liver depuration of LDLs and an increase in apolipoprotein B (ApoB) and very low-density lipoprotein (VLDL) synthesis.2 High-density lipoprotein (HDL) particle formation is mediated by the quantity of cholesterol contained in the endosomes/lysosomes and by the ATP-binding cassette transporter A1 (ABCA1). Altered regulation of that transporter causing a decrease in HDL particle formation has been found in studies on fibroblasts in LAL-D patients.14–16
Those mechanisms explain the dyslipidemia present in LAL-D patients: elevated total cholesterol (TC) and LDL-cholesterol (LDL-C), low concentrations of HDL-cholesterol (HDL-C), and hypertriglyceridemia.15
Clinical manifestationsThe diagnosis of LAL-D is made through the clinical history, intentionally looking for its signs and symptoms, and complementing it with the corresponding laboratory tests.6,17
The early clinical manifestations of the disease are diverse and hyporexia, frequent vomiting, diarrhea, steatorrhea, malnutrition and failure to grow, and hepatosplenomegaly stand out among them.2,3
Liver damage rapidly progresses to portal hypertension and severe liver dysfunction in the first months of life. Metabolic acidosis and adrenal gland calcification have been described in almost one third of the patients.2,10
Severe liver dysfunction is early and progressive in LAL-D presenting in infancy, or Wolman disease.
Clinical presentation varies in children and adults. The mean age for symptom onset is 5 years, but some patients remain asymptomatic and are not diagnosed until adulthood. Abdominal distension from visceromegaly and short stature are common.1
Clinical manifestations in late-presentation LAL-D can be nonspecific, such as abdominal distension and short stature.
Abdominal visceromegaly is frequent in LAL-D, and when detected, the disease should be suspected.
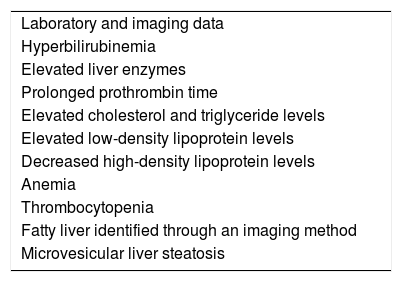
In addition to the clinical manifestations of chronic liver disease and its complications, such as portal hypertension, bleeding, advanced-stage encephalopathy, and hepatocellular carcinoma, LAL-D manifestations also include: abdominal pain, diarrhea with or without malabsorption, gallbladder disease with cholelithiasis, and repeat epistaxis due to thrombocytopenia.1,15Table 1 lists the main clinical findings in late-presentation LAL-D and Table 2 lists the laboratory test results.2,18,19
Main laboratory data of late-presentation LAL-D.
| Laboratory and imaging data |
| Hyperbilirubinemia |
| Elevated liver enzymes |
| Prolonged prothrombin time |
| Elevated cholesterol and triglyceride levels |
| Elevated low-density lipoprotein levels |
| Decreased high-density lipoprotein levels |
| Anemia |
| Thrombocytopenia |
| Fatty liver identified through an imaging method |
| Microvesicular liver steatosis |
Late-presentation LAL-D should be ruled out in patients with hepatosplenomegaly, persistent aminotransferase elevation, and dyslipidemia.
The accidental finding of aminotransferase elevation requires a search for liver and spleen enlargement.
DyslipidemiaThe primary cardiovascular manifestations are: coronary artery disease, aneurysm, and cerebrovascular accident.1,20 Complications related to early-onset atherosclerotic cardiovascular disease are associated with dyslipidemia secondary to hepatic alterations of lipid metabolism.21 Different studies have reported that complications associated with early-onset atherosclerotic cardiovascular disease present in 4.2% of patients with LAL-D and in 12.5% of patients with LAL-D above 25 years of age.15,22,23
Dyslipidemia associated with late-presentation LAL-D is related to atherosclerosis and is a risk factor for early cardiovascular disease.
Dyslipidemia in children and adults with late-presentation LAL-D is characterized by elevated concentrations of total cholesterol, LDL, and ApoB and by low levels of HDL. Some cases can present with hypertriglyceridemia.
Laboratory work-upBiochemically, the majority of patients with LAL-D present with elevated levels of alanine aminotransferase and aspartate aminotransferase, mild hyperbilirubinemia, elevated levels of gamma-glutamyl transferase, prolonged prothrombin time, prolonged fasting hypoglycemia, and IIA or IIB dyslipidemia, with hypercholesterolemia, low levels of HDL-C, elevated levels of LDL-C, Apo B, and sometimes hypertriglyceridemia.24 LDL concentrations are generally higher than those in patients with nonalcoholic fatty liver disease, with mean concentrations of 100-200mg/dL, which is not as high as that of patients with heterozygote familial hypercholesterolemia. Anemia is identified in 12.5% of cases, and thrombocytopenia and leukopenia are found in patients that present with hepatosplenomegaly or cirrhosis of the liver. Elevated levels of alanine aminotransferase have been observed in 91.7%, of aspartate aminotransferase in 76.2%, of gamma-glutamyl transferase in 20%, and of total bilirubin in 20%.15
A complete lipid profile and periodic monitoring of aminotransferases for at least six months are recommended in all patients with liver disease.
Imaging studiesAdrenal gland calcification is identified through plain x-ray in 50% of children with early-onset disease presentation.1,2
Adrenal gland calcification is characteristic of LAL-D that presents in infancy.
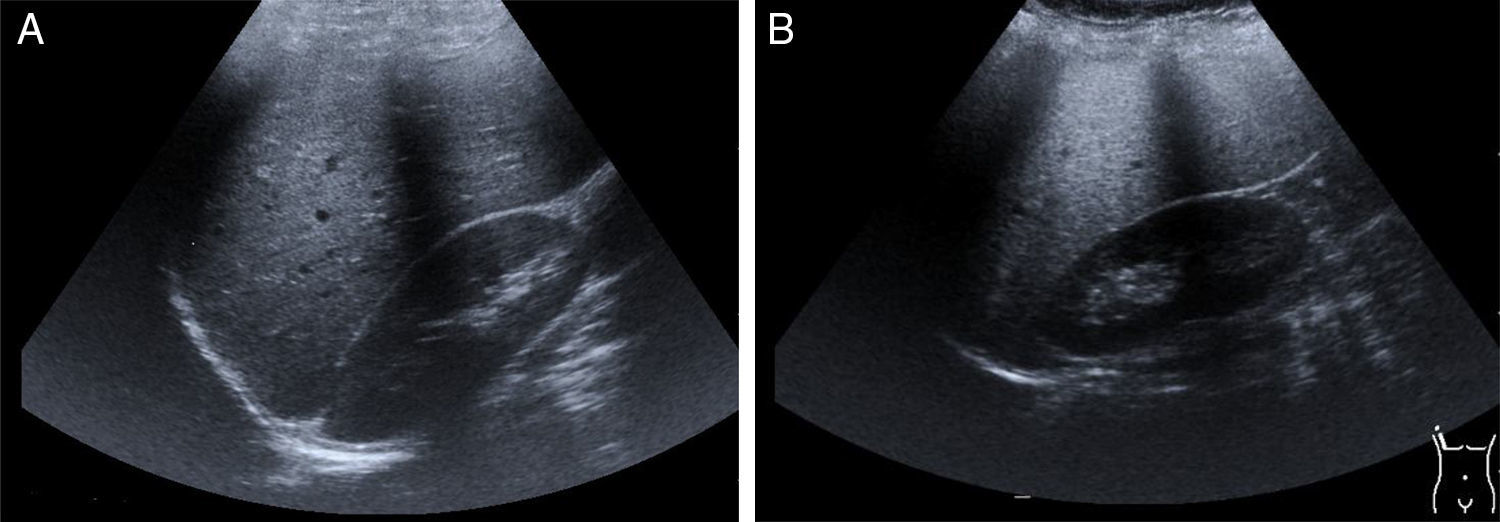
Hepatomegaly and splenomegaly are corroborated through imaging studies and they also identify signs of liver steatosis. Ultrasound, computed axial tomography, spectroscopy magnetic resonance, and transition elastography or the fusion of elastography and magnetic resonance, are noninvasive diagnostic tools25 for evaluating and staging fatty liver (fig. 2A and2B).
 Ultrasound image of a normal liver.")
Late-presentation LAL-D is a cause of liver steatosis, identifiable through ultrasound imaging.
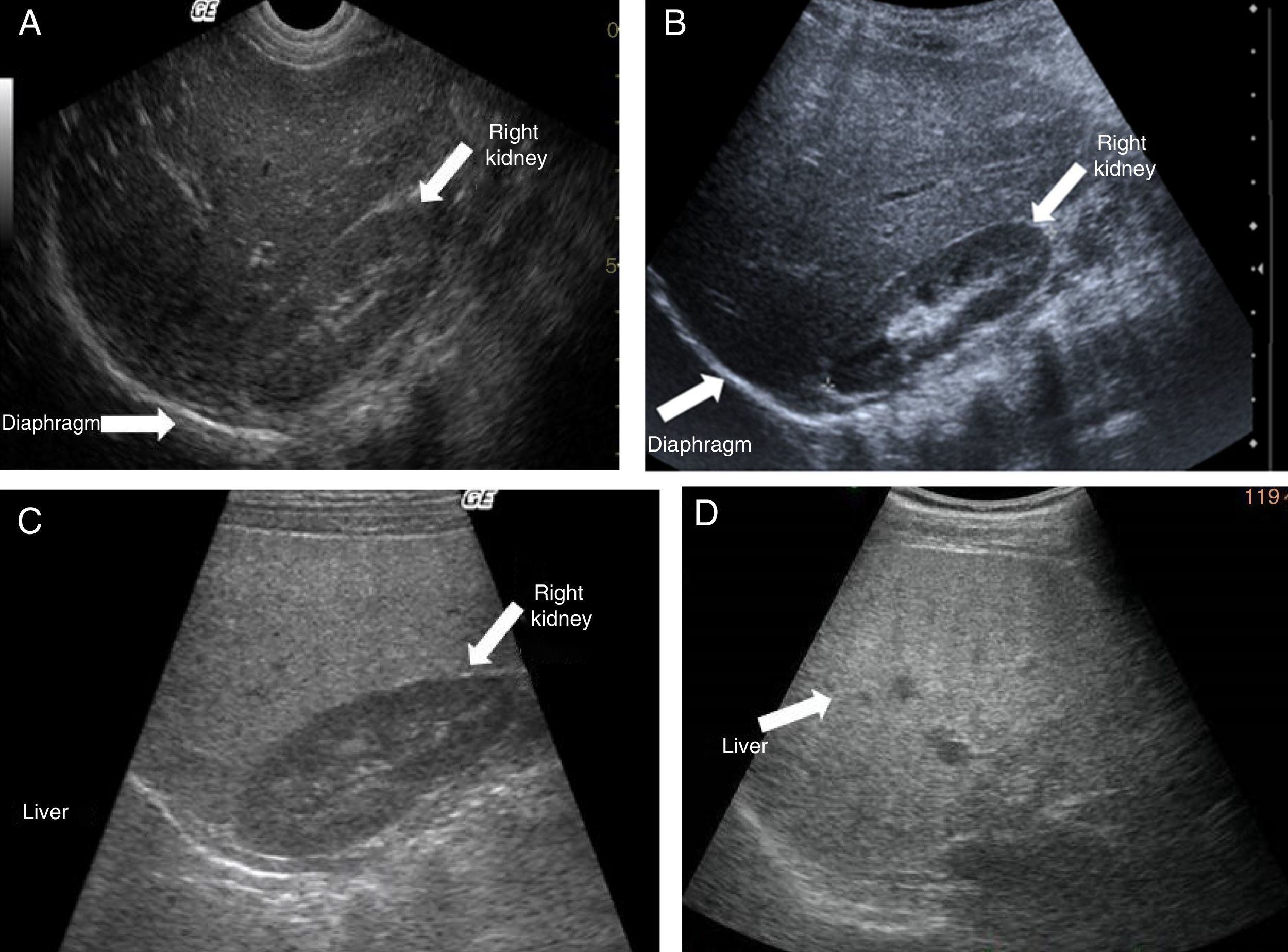
Ultrasound shows diffuse echogenicity and identifies hepatomegaly and splenomegaly (fig. 3A). The Tominaga classification26 for evaluating liver echogenicity includes three stages: a) mild, with a minimal diffuse increase in liver echogenicity, normal aspect of the diaphragm and intrahepatic vessels (fig. 3B), b) moderate, modest diffuse increase in liver echogenicity, slight decrease in intrahepatic vessels and diaphragm (fig. 3C), and c) severe, marked increase in echogenicity, poor penetration of the posterior segment of the right hepatic lobe and poor or null identification of the hepatic vessels and diaphragm (fig. 3D).
 Ultrasound image showing conserved echogenicity in the liver and right kidney.")
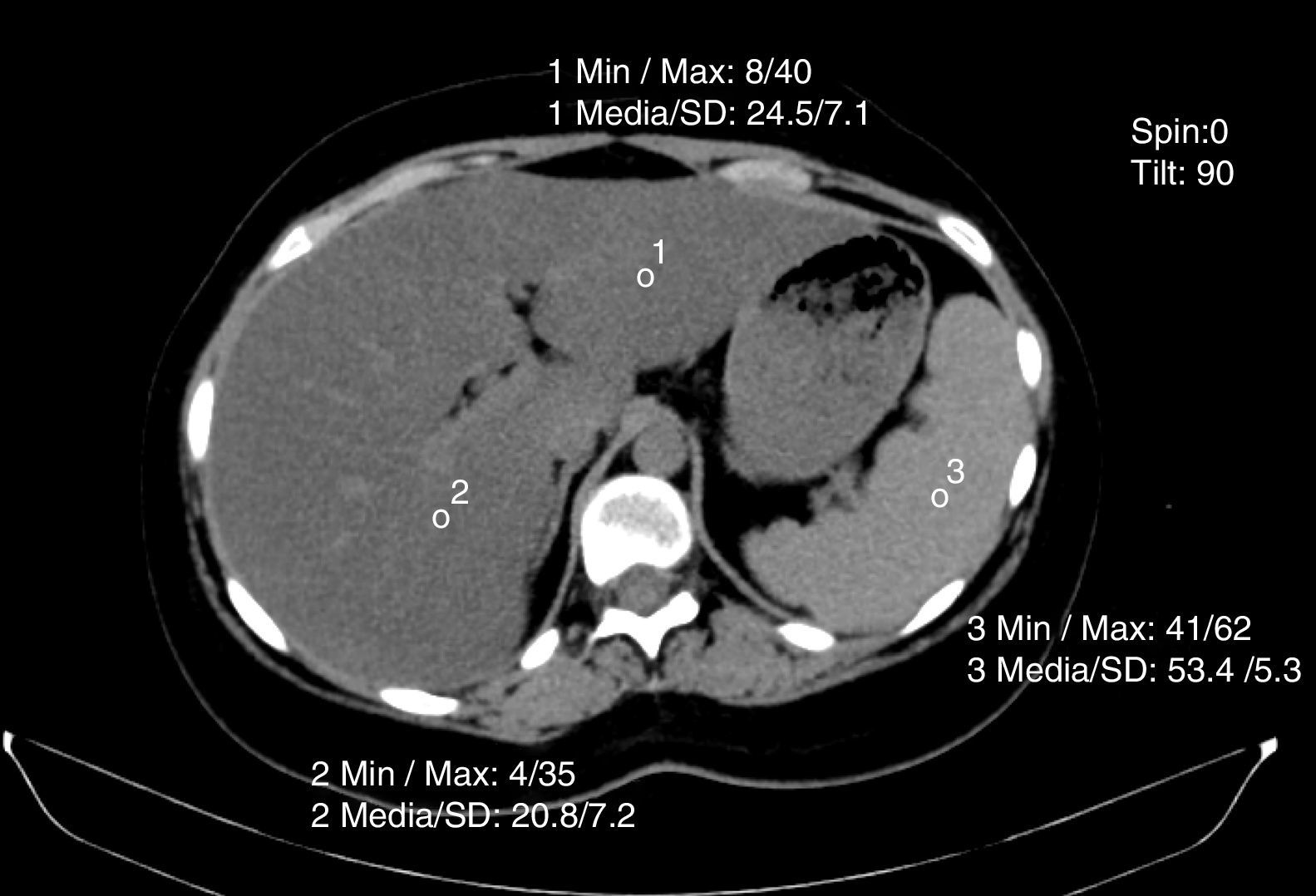
Non-contrasted computed axial tomography evaluates liver attenuation measured in Hounsfield units and attenuation values between the liver-spleen.27 If those values are below 48 Hounsfield units, fat infiltration is diagnosed (fig. 4). Magnetic resonance and elastography optimization are also diagnostic methods, as is nuclear magnetic resonance spectroscopy.

Hydrogen (1H) and carbon (13C) isotope determination in fatty tissue differs in patients with LAL-D, compared with patients with NAFLD, because of the methyl group (CH3) to methylene group (CH2) molar ratio. A cholesterol moiety has five CH3 groups with a strong resonance on the 1H magnetic resonance spectrum compared with a fatty acid moiety that has one methyl group per molecule.28 In LAL-D patients, the mean concentration of cholesterol in liver tissue was 21.1±13.6mM and the mean concentration of fatty acids was 51.2±19.2mM, whereas in NAFLD patients the mean fatty acid concentration was 442±164mM.28 That signifies a correlation between biochemical determination and cholesterol and fatty acid determination through magnetic resonance spectroscopy, which could be a useful, noninvasive alternative for determining cholesterol and triglycerides in the liver and monitoring disease progression.
PathologyThe histopathologic study of biopsies from patients with LAL-D offers at least three benefits: a) on its own, it can be indicative of the disease, b) it contributes to confirming diagnosis when the disease is suspected, and c) it provides valuable information for establishing outcome and is useful in patient follow-up, treatment evaluation, and for a retrospective analysis.29
Histopathologic alterations in LAL-D are characteristic and vary in intensity according to age, length of progression, and disease expressivity. The liver and all organs with a predominance of macrophages are the most commonly affected.1,7 In LAL-D that presents in infancy there are florid manifestations, not only in the liver, but also in the different organs that are extensively affected.1
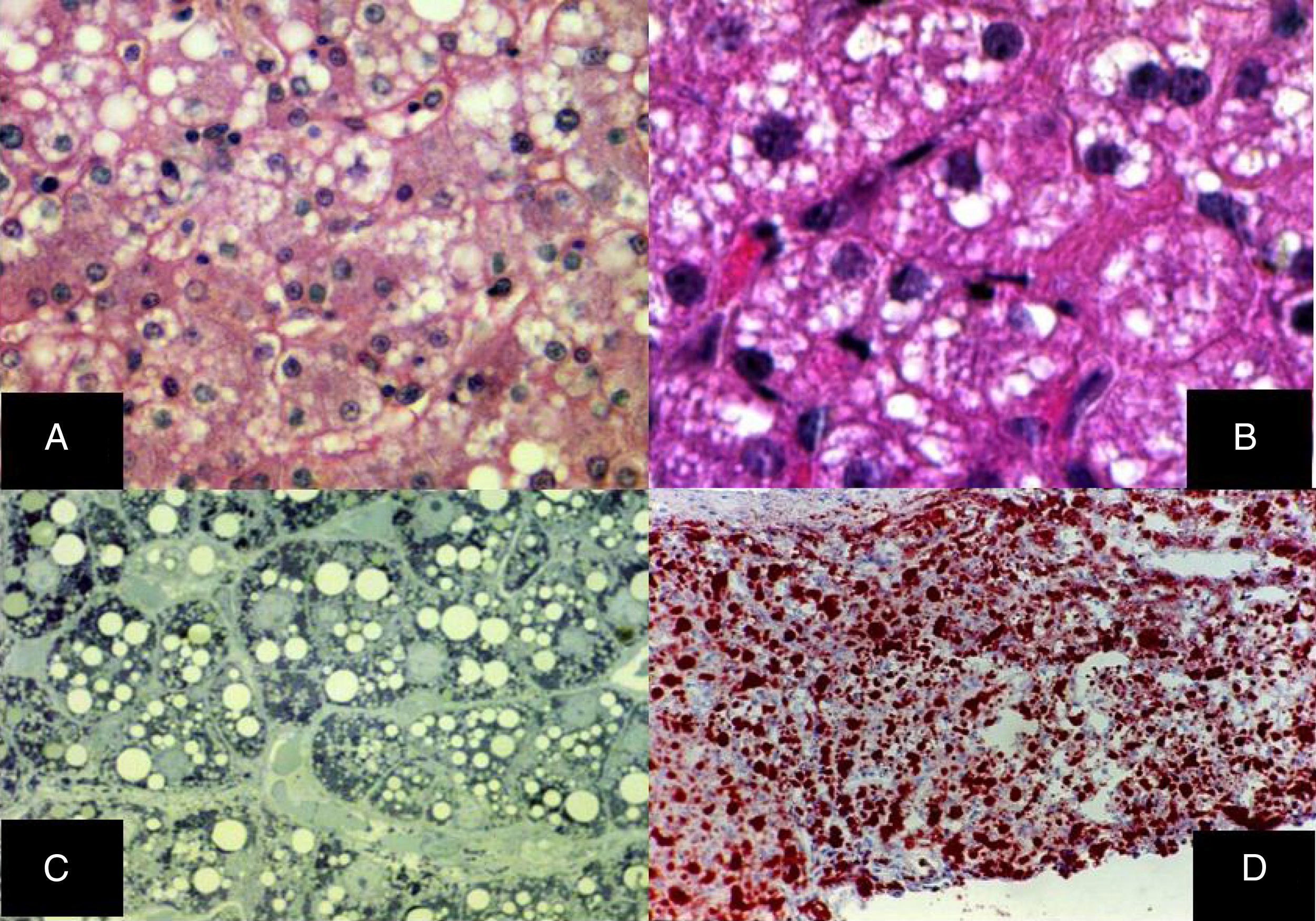
Diffuse microvesicular steatosis is the basic lesion in the liver. The cytoplasm of the hepatocytes contains numerous lipid droplets that are intensely positive with the oil red O stain. Those vesicles measure less than one micron in diameter and do not displace the nucleus (fig. 5).29–32
 Diffuse microvesicular steatosis with numerous clear vacuoles inside the cytoplasm of the hepatocytes (H&E, original magnification x400). B) The vacuoles measure around one micron in diameter and do not displace the nucleus (H&E, original magnification x400). C) Semi-fine slices that show the microvesicular nature of the steatosis, seen in practically all the hepatocytes (toluidine blue, original magnification x400). D) Multiple neutral fat droplets are revealed in the entire hepatic lobule (oleous red, original magnification x200).")
A) Diffuse microvesicular steatosis with numerous clear vacuoles inside the cytoplasm of the hepatocytes (H&E, original magnification x400). B) The vacuoles measure around one micron in diameter and do not displace the nucleus (H&E, original magnification x400). C) Semi-fine slices that show the microvesicular nature of the steatosis, seen in practically all the hepatocytes (toluidine blue, original magnification x400). D) Multiple neutral fat droplets are revealed in the entire hepatic lobule (oleous red, original magnification x200).
Diffuse microvesicular steatosis and cholesterol crystals in the cytoplasm of hepatocytes and macrophages are liver biopsy findings suggestive of LAL-D.
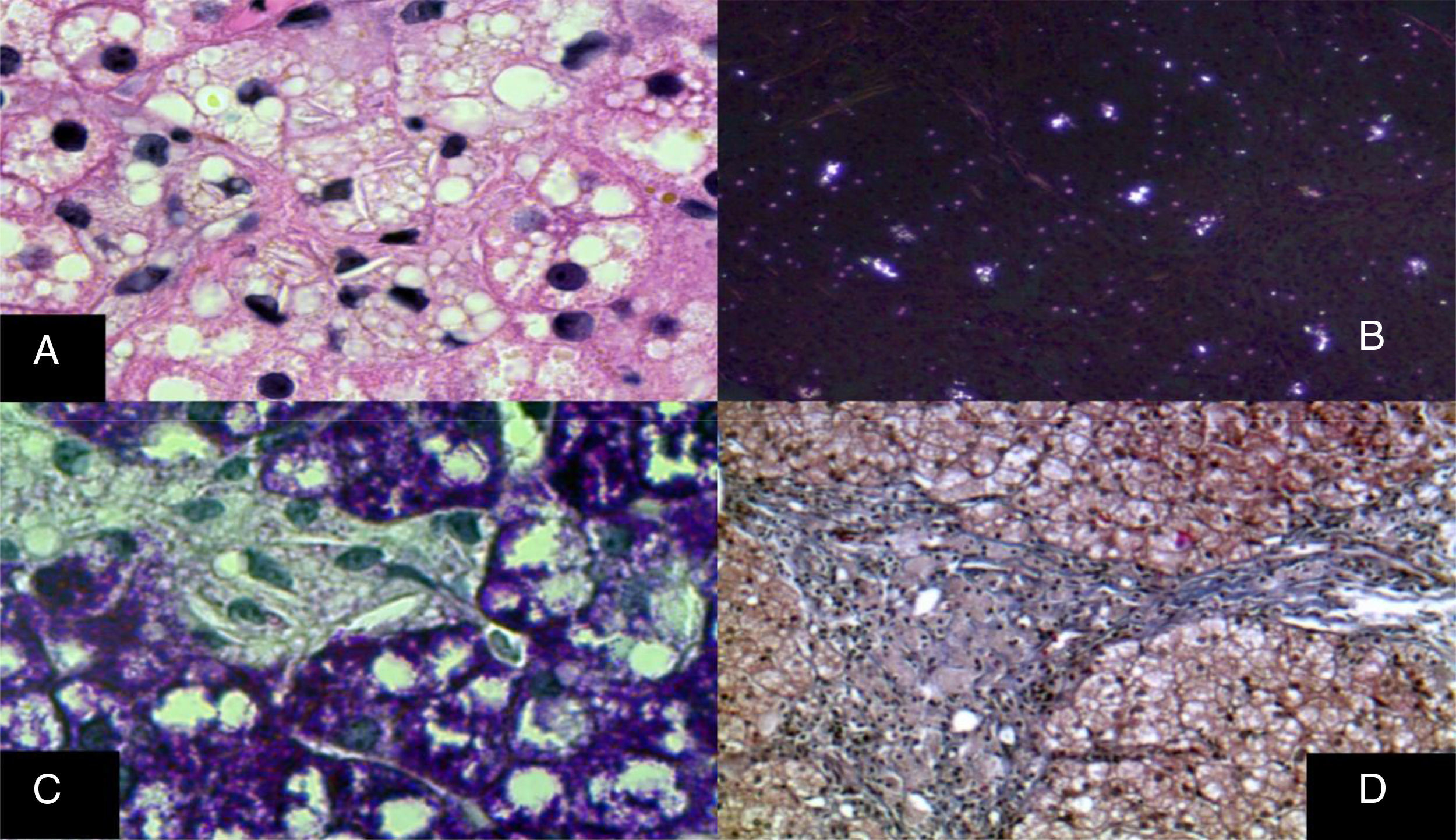
The monocyte-macrophage system lesion affects Kupffer cells and the portal macrophages. Both show small droplets and cholesterol crystals in their cytoplasm. In addition, macrophages accumulate pigment similar to lipofuscin (fig. 6).29–33 Hepatocytes can have cholesterol crystals in their cytoplasm that exhibit a characteristic silver birefringence when viewed under polarized light.29–33
 Immunohistochemical reaction showing that in situ in both hepatocytes and Kupffer cells clear grooves are seen in their cytoplasm, corresponding to cholesterol crystals (H&E, original magnification x1,000). B) Silver birefringence characteristic of the numerous cholesterol crystals (polarized light, original magnification x100). C) Kupffer cells with microvesicular steatosis and cholesterol crystals clearly stand out (PAS, original magnification x1,000). D) A porta hepatis space expanded by numerous macrophages with “foamy” cytoplasm due to lipid inclusions (Masson's trichrome, original magnification x200).")
A) Immunohistochemical reaction showing that in situ in both hepatocytes and Kupffer cells clear grooves are seen in their cytoplasm, corresponding to cholesterol crystals (H&E, original magnification x1,000). B) Silver birefringence characteristic of the numerous cholesterol crystals (polarized light, original magnification x100). C) Kupffer cells with microvesicular steatosis and cholesterol crystals clearly stand out (PAS, original magnification x1,000). D) A porta hepatis space expanded by numerous macrophages with “foamy” cytoplasm due to lipid inclusions (Masson's trichrome, original magnification x200).
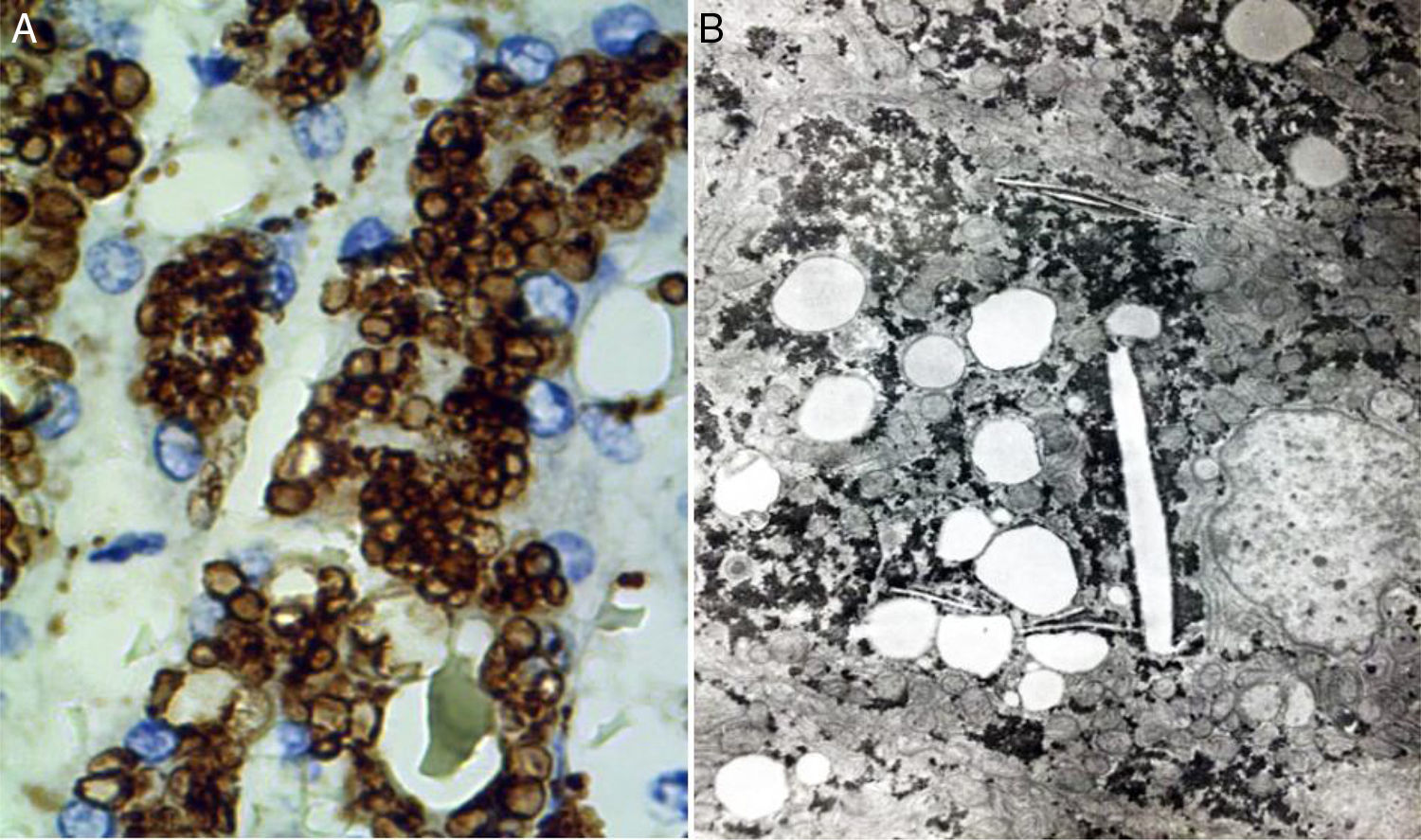
Two basic tools demonstrate the existence of lipids inside lysosomes: immunohistochemistry and electron microscopy. Antibodies can be used that recognize lysosomal components, such as lysosomal integral membrane protein-2 (LIMP2), a protein associated with lysosome associated membrane protein-1 (LAMP1), and the lysosomal luminal protein or cathepsin D (fig. 7A).
 Immunohistochemical reaction showing lipid droplets inside lysosomes (cathepsin D, original magnification x400). B) Photomicrograph showing lipids surrounded by simple lysosomal membrane that reveals its lysosomal nature, as well as cholesterol crystals (electron microscopy, original magnification x2,000).")
A) Immunohistochemical reaction showing lipid droplets inside lysosomes (cathepsin D, original magnification x400). B) Photomicrograph showing lipids surrounded by simple lysosomal membrane that reveals its lysosomal nature, as well as cholesterol crystals (electron microscopy, original magnification x2,000).
Through electron microscopy, lipids are viewed inside the lysosomes (fig. 7B), surrounded by a simple membrane unit and not freely accumulated in the cytosol. Liver fibrosis is dependent on progression time and disease expressivity, which can range from mild disease to cirrhosis.29,31
Late-presentation LAL-D should be ruled out through immunohistochemistry or electron microscopy in patients with biopsy-demonstrated steatosis that is predominantly microvesicular
With that histopathologic data and the demonstration of lipid storage in the lysosomes, LAL-D diagnosis can be made, differentiating it from other entities that present with microvesicular steatosis, including other storage diseases,29,30 as shown in Table 2.
Late-presentation LAL-D should be ruled out in non-obese adult patients with cryptogenic cirrhosis of the liver.
Enzymatic and molecular diagnosis of LAL-DLAL (E.C.3.1.1.13), also known as cholesteryl ester synthase, cholesterol esterase, triterpenol esterase, cholesteryl esterase, lipase A, and steryl-ester acylhydrolase, is the enzyme in charge of hydrolyzing the cholesteryl esters and triglycerides of low-density lipoproteins, producing cholesterol and free fatty acids.2 Therefore, its deficiency leads to lipid accumulation and alterations in lipid homeostasis.34,35
The biochemical diagnosis of LAL-D in leukocytes, fibroblasts, and amniotic fluid has been in use for 50 years36,37 and in 2012, an inhibitor (Lalistat 2) was incorporated in the technique that improved its specificity.38–40
The dried blood spot (DBS) test on filter paper can be used on patients of any age and it enables LAL activity to be determined. It is useful for detecting affected patients2 and its sensitivity and specificity is very high.38 A small amount of blood is obtained through puncture and immediately placed on Whatman 903 filter paper. The blood is absorbed and left to dry at room temperature until the following day. The sample should not be exposed to heat and should be transported in double bags with desiccant, to maintain enzyme activity. It can be stored long-term at -20°C.41,42 Normal LAL activity values depend on the laboratory where they are processed. An international expert consensus for LAL diagnosis determined the importance of analyzing LAL activity results based on the percentage of activity, with respect to the normal mean of the healthy reference population analyzed by each laboratory. Affected patients are those with an enzyme activity value below 5%, patients in the gray zone have values between 5 and 10%, and non-affected patients have activity > 10%, in relation to the mean value of the healthy population.43 The advantages are: a small amount of blood is taken, blood sample collection is easy, there is adequate stability under room temperature conditions, it facilitates rapid diagnosis in large populations, management is easy, and the cost is low.37,38,41,42
In Mexico, the dried blood spot test is carried out at the gastroenterology laboratory of the Hospital Infantil de México Federico Gómez and the Centro de Investigación Biomédica de Occidente in Guadalajara.
Enzymatic diagnosis of LAL-D can be made through the dried blood spot test on filter paper.
Molecular diagnosis of LAL-D requires sequencing of the LIPA gene. It is located at 10q23.31, contains 10 exons, measures 36kb, and produces a protein made up of 399 amino acids. The test is carried out in peripheral blood leukocytes and involves the sequencing of the LIPA gene or of a gene panel containing it. DNA is extracted from the sample (leukocytes or DBS) for sequencing the gene or for massive sequencing.9,40,44,45
When LAL-D diagnosis is not confirmed through gene or gene panel sequencing, then the genomic approach should be considered.40 At present more than 40 mutations have been detected in the LIPA gene,1,40,46–49 and the most frequent (50%-70%) is located at the exon 8 splice junction (E8SJM1G>A). In Mexico, the c.253C>A and c.294C>G mutations have been found in patients with the CESD phenotype. Molecular diagnosis should be considered as a support method, given that the mutation(s) responsible cannot always be identified, as well as its high cost. Enzymatic activity determination on paper is an accessible, accurate, and less costly test.
Differential diagnosisBecause of its variability, LAL-D should be differentiated at its neonatal stage from the different causes of neonatal cholestasis. Metabolic diseases clearly stand out as such causes, especially those that present with rapid-progression liver disease, such as tyrosinemia, neonatal galactosemia, hemochromatosis, and Niemann-Pick disease type C.50–53
LAL-D should be considered in the differential diagnosis of neonatal cholestasis with liver failure.
Because type IIA or IIB hyperlipidemia has been observed in patients with LAL-D, heterozygous familial hypercholesterolemia, defective familial ApoB 100, combined familial hyperlipidemia, and polygenic hypercholesterolemia should be considered in the differential diagnosis.11,54,55
Familial hypercholesterolemia can be confused with LAL-D, given that LDL is elevated in both diseases, but there is no hepatosplenomegaly in the former. Familial hypercholesterolemia is an autosomal co-dominant inherited disease, whereas LAL-D is an autosomal recessive inherited disease.56,57
Late-presentation LAL-D should be considered in the differential diagnosis in all children and adults suspected of having heterozygous familial hypercholesterolemia.
Because patients with LAL-D present with hepatomegaly, elevated levels of liver enzymes, fatty liver, and dyslipidemia, the differential diagnosis should include metabolic syndrome, which has a greater risk for cerebrovascular disease and type 2 diabetes mellitus.58 Only 27.1% of patients with LAL-D present with hypertriglyceridemia, but they are not obese.2,15
Late-presentation LAL-D should be considered in non-obese individuals with or without metabolic syndrome that present with hepatomegaly, elevated aminotransferase levels, fatty liver, and dyslipidemia.
ConclusionLAL-D is a little-known, and consequently unsuspected disease that can go unnoticed or be confused with other pathologies and consequently misdiagnosed. The algorithm proposed herein provides a sequence of actions to be taken, based on the clinical manifestations collected from different studies to optimize the diagnostic process of patients with LAL-D.
AlgorithmGiven the heterogeneous phenotype of LAL-D, a patient does not need to meet all the criteria previously described to consider the disease within the differential diagnosis. The criteria for LAL-D suspicion are put forward, taking two large groups into account: that which presents with any entity that affects the liver, and that which manifests with symptoms of dyslipidemia/atherosclerosis/early cardiovascular disease. Both groups can be accompanied with or have a history of chronic diarrhea, short stature, anemia, gallstones, or bleeding.
In the group of patients with liver involvement with no clear etiology, LAL-D diagnosis should be considered and the dried blood spot test on filter paper should be performed to determine LAL in blood.
In the group that primarily presents with dyslipidemia/atherosclerosis/early cardiovascular disease, dyslipidemia should be identified as IIA or IIB. When there is an increase in LDL-C and a decrease in HDL-C, reference values for the pediatric population should always be considered. However, it should be kept in mind that normal LDL-C and below normal HDL-C values can be found, especially in patients with liver disease. Therefore, aminotransferase determination is recommended and if they are high, LAL determination through the dried blood spot test on filter paper should be performed.
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Vázquez-Frias R, García-Ortiz JE, Valencia-Mayoral PF, Castro-Narro GE, Medina-Bravo PG, Santillán-Hernández Y, et al. Consenso mexicano sobre el diagnóstico de la deficiencia de lipasa ácida lisosomal. Revista de Gastroenterología de México. 2018;83:51–61.

