
An 18-year-old male was referred to us due to barium study findings describing a 15mm sessile polyp in the ileum.
His father had been operated on for polyps of the colon and small bowel that were never studied. When the patient was 9 years old, he presented with an episode of hematochezia secondary to polyps of the colon that were endoscopically resected, but with no follow-up.
When referred to us, the patient was asymptomatic with no abdominal pain or bowel rhythm alteration. Physical examination revealed millimetric-sized pigmented macules on the lower lip and fingers with no other alterations.
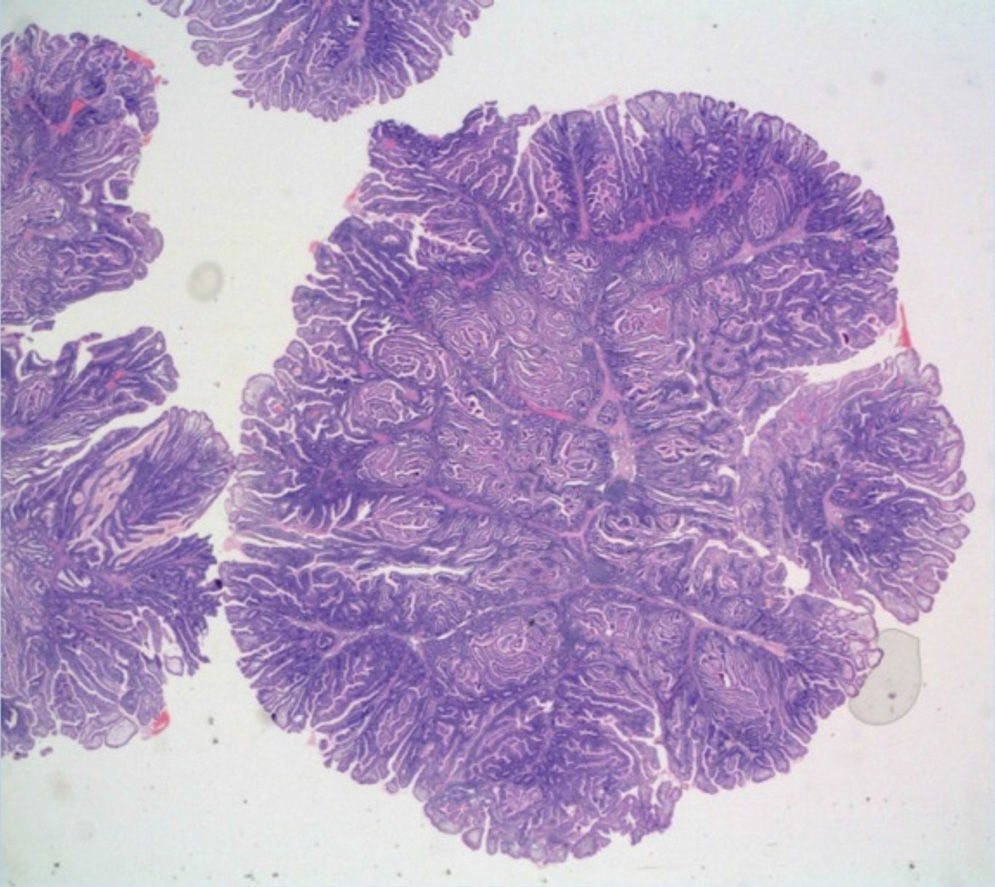
One month later, a polypoid lesion was confirmed through capsule endoscopy and ileoscopy. Endoscopic exeresis was not possible and so ileocolic resection was carried out, resulting in the finding of hamartomatous polyps with no dysplasia (fig. 1).

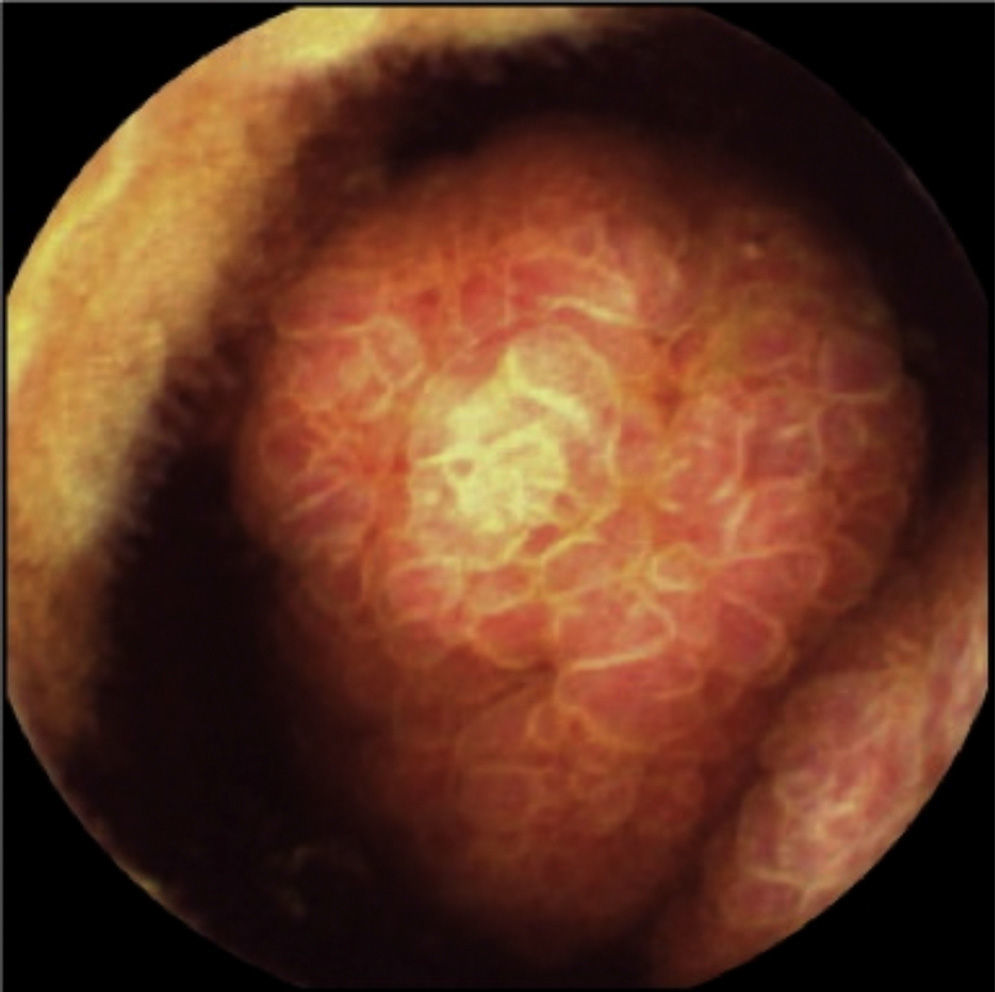
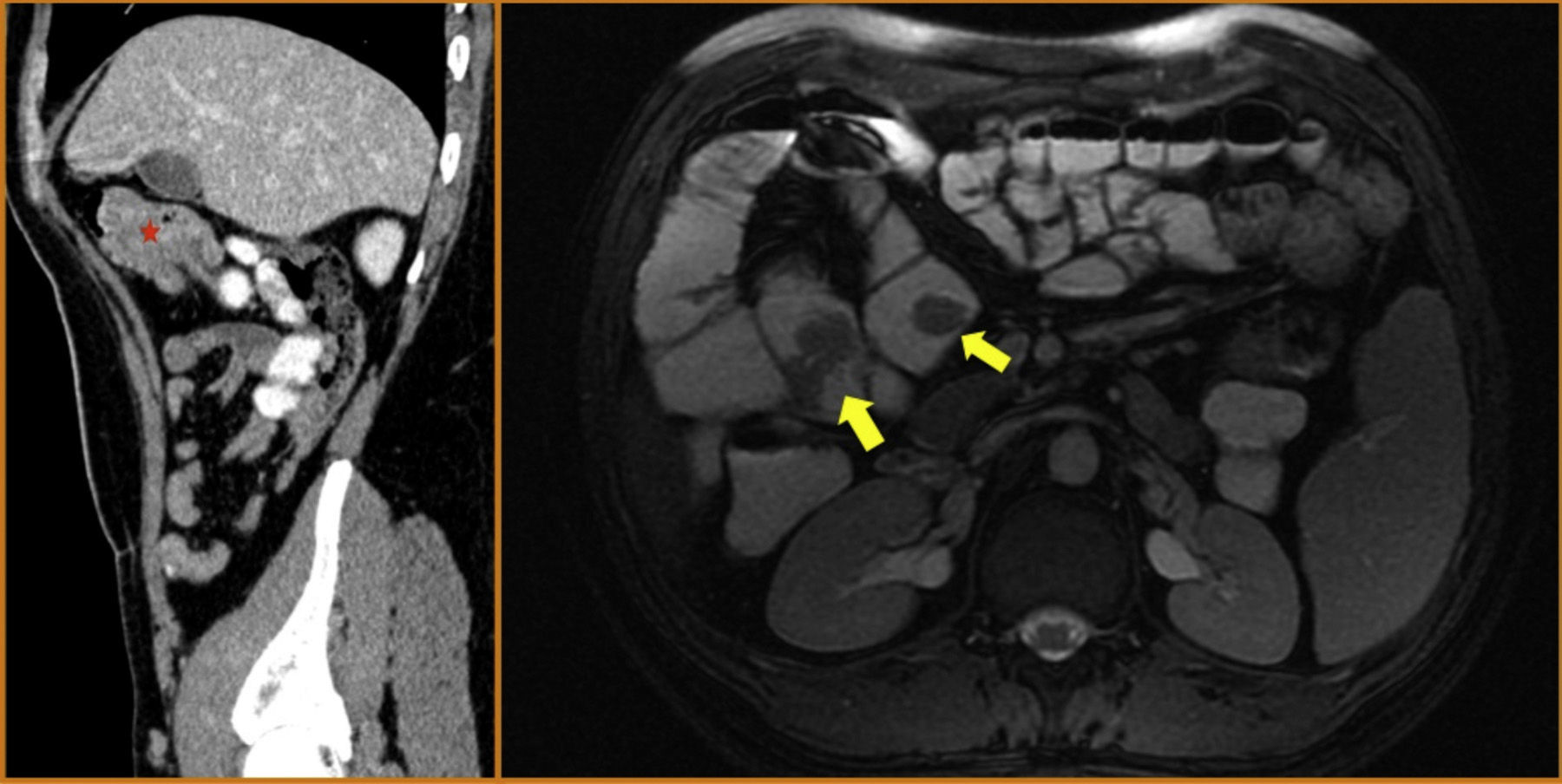
The patient remained asymptomatic during the follow-up with resection of multiple colonic and gastric polyps. However, the last capsule endoscopy, performed 2 months after surgery, revealed a large ulcerated polyp in the jejunum occupying the entire intestinal lumen (fig. 2). These findings were confirmed through magnetic resonance enterography and computerized axial tomography, which identified various polypoid lesions in the jejunum that were acting as the head of the intussusception of a bowel segment (fig. 3).

. On the right, the axial magnetic resonance enterographic image identifies several polypoid lesions in the jejunum and proximal ileum (yellow arrows).")
On the left, the sagittal CT image shows various polypoid formations that are acting as the head of the intussusception of a jejunal segment (asterisk). On the right, the axial magnetic resonance enterographic image identifies several polypoid lesions in the jejunum and proximal ileum (yellow arrows).
Two months later, after a failed attempt to reach the lesion through push enteroscopy, double-balloon enteroscopy was performed. The polyp described was reached via oral, but endoscopic resection was impossible, making it necessary for the patient to undergo surgery again one month later. Through various enterotomies, 10 hamartomatous polyps were resected, some of which were in an invaginated segment.
With these clinical and histopathologic data, Peutz-Jeghers syndrome (PJS) was diagnosed. A genetic study was requested in which complete sequencing of the STKB11 (LKB1) gene was carried out, confirming that the patient was a carrier of a pathogenic mutation in exon 4 (p.Asp194Asn). The patient has remained asymptomatic after surgery and has had no relevant findings in the surveillance tests.
PJS is a rare genetic disease with dominant autosomal heredity caused by a germ cell mutation in the STK11 (LKB1) gene located on the 19p13.3 chromosome. It has an estimated incidence of one out of every 50,000-200,000 births.1 Precisely because of the low frequency of this syndrome, there are no well established protocols in relation to surveillance and management.
PJS diagnosis is based on the presence of some of these criteria:2
- –
If there is no family history of PJS:
- •
Two or more hamartomatous polyps histologically confirmed, or
- •
Any number of hamartomatous polyps with the characteristic mucocutaneous pigmentation
- •
- –
If there is a family history of PJS, any number of hamartomatous polyps or mucocutaneous pigmentation
The cutaneous lesions appear in 95% of the patients and consist of dark brown-black pigmented macules that are most frequently located in the perioral region (94%), on the hands (74%), and in the buccal mucosa (66%).3
The large majority of patients will present with hamartomatous polyps. They tend to be more frequently located in the small bowel (60-90%), followed by the colon (50-64%), stomach (49%), and rectum (32%), and can also be extraintestinal. Number and size is quite variable and they have no specific endoscopic characteristics.4
To make the adequate differential diagnosis from the other types of polyposis, we must be familiar with the 3 types of polyps that are each associated with a specific syndrome. Among the adenomas, classic or attenuated familiar adenomatous polyposis or polyposis associated with the MYH gene are the most frequent syndromes. Serrated polyposis is associated with serrated polyps. And finally, in addition to PJS, juvenile polyposis, Cowden syndrome, and Bannayan-Riley-Ruvalcaba syndrome are among the types of hamartomatous polyposis, each with different clinical and extraintestinal manifestations.5
The most frequent clinical onset of PJS is bowel obstruction (43%), followed by abdominal pain of ischemic origin (23%), rectorrhagia (14%), or polyp extrusion through the rectum (7%).1 Up to 69% of the patients can present with some intussusception phenomenon produced by polyps >15mm, and thus the resection of polyps of those sizes could avoid such events.6
There is also an increased risk in these patients for tumors. The most frequent neoplasias are those of the colon, followed by breast and small intestine tumors. The accumulative risk increases with age and varies from 37 to 93% with a relative risk of 9.9-18.7
Surveillance of these patients should be directed at the risk for intussusception and neoplasias, but no screening protocols have been validated in clinical trials. There is a guide establishing some recommendations that include not only the endoscopic surveillance of gastrointestinal polyps, but also screening for extraintestinal neoplasias such as breast or testicular cancers.8 The international group for the screening of cancer of the pancreas has recently included patients with PJS among the high risk patients, regardless of whether they have a family history of pancreatic cancer.9
There is no consensus on how to treat polyps when they are detected, but even though the detection of dysplasia in hamartomas is low, endoscopic polypectomy is safe, given that emergency surgery is avoided and it protects the patient from developing intestinal and extraintestinal neoplasias.10
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Adán-Merino L, Aldeguer-Martínez M, Lozano-Maya M, Hernández-García-Gallardo D, Casado-Fariñas I. Luces y sombras en el diagnóstico y seguimiento de un paciente joven asintomático con síndrome de Peutz-Jeghers. Revista de Gastroenterología de México. 2016;81:59–61.

